Tränendrüsentumoren sind Neoplasien, die von der Tränendrüse in der Orbita ausgehen. Die jährliche Inzidenz beträgt etwa 1 pro 1 Million Personen, und sie machen etwa 10 % aller raumfordernden Orbitaprozesse aus.
Die Tränendrüse teilt embryologisch ihren Ursprung mit den Speicheldrüsen, daher wird für die Tumorklassifikation das Klassifikationssystem der Speicheldrüsentumoren verwendet.
Tränendrüsentumoren werden grob in epitheliale Tumoren und nicht-epitheliale Tumoren eingeteilt. Die folgende Tabelle zeigt die wichtigsten Klassifikationen und Merkmale.
Pleomorphes Adenom: Häufigster benigner Tumor, der etwa 70 % der epithelialen Tränendrüsentumoren ausmacht. Histologisch zeigt es ein vielgestaltiges Bild mit einer Mischung aus epithelialen und stromalen Gewebekomponenten. Tritt häufiger bei Frauen im Alter von 30–40 Jahren auf.
Adenoidzystisches Karzinom: häufigster epithelialer maligner Tumor (ca. 60%). Tritt häufiger bei Männern auf, vom Jugend- bis ins hohe Alter.
Plomorphes Adenokarzinom: maligne Transformation innerhalb eines pleomorphen Adenoms.
Mukoepidermoidkarzinom: unterteilt in niedrig- und hochmaligne.
Primäres Adenokarzinom: sehr schlechte Prognose (3-Jahres-Mortalität 70%).
Die jährliche Inzidenz okulärer Adnexlymphome steigt um 4,5 %. 1)
QWelche Arten von Tränendrüsentumoren gibt es?
A
Sie werden grob in epitheliale und nicht-epitheliale Tumoren eingeteilt. Bei den epithelialen sind das benigne pleomorphe Adenom und das maligne adenoidzystische Karzinom typisch, bei den nicht-epithelialen überwiegen maligne Lymphome (insbesondere MALT-Lymphome). Siehe die Klassifikationstabelle in diesem Abschnitt für Details.
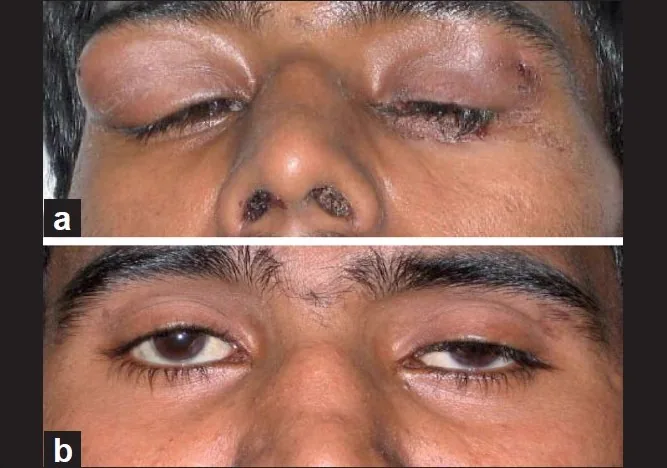
Khanna D, et al. Suppurative dacroadenitis causing ocular sicca syndrome in classic Wegener’s granulomatosis. Indian J Ophthalmol. 2011. Figure 1. PMCID: PMC3116546. License: CC BY.
(a) Bilaterale Dakryoadenitis mit nasalen Krusten und (b) Verschwinden des Tränendrüsentumors einen Monat nach Immunsuppression, obwohl eine linksseitige Ptosis sichtbar ist. Entspricht der Tränendrüsenschwellung, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Die Art der Symptome variiert je nach Tumorart, Wachstumsgeschwindigkeit und Malignität.
Schwellung des Oberlids: Eine Schwellung mit einer Raumforderung ist das typische Erstsymptom.
Exophthalmus: Vorwärtsverlagerung des Augapfels durch die Raumforderung.
Ptosis: Herabhängen des Oberlids. Beim pleomorphen Adenom ist dies in etwa der Hälfte der Fälle das Erstsymptom.
Doppelbilder: Treten häufig beim Blick nach oben-außen auf. Bei langsam wachsenden Tumoren kann dies unbemerkt bleiben.
Schmerzen: Treten bei schnell wachsenden Tumoren, entzündlichen Läsionen und adenoid-zystischen Karzinomen auf. Das pleomorphe Adenom ist in der Regel schmerzlos.
Die klinischen Befunde sind je nach Tumorart charakteristisch.
Pleomorphes Adenom
Augapfelverlagerung: Eine Verlagerung nach vorne, unten oder nasal unten ist typisch.
Langsames Wachstum: Die durchschnittliche Symptomdauer beträgt etwa 2 Jahre. Charakteristisch ist die Schmerzlosigkeit.
Tastbarer subkutaner Tumor: Am temporalen Oberlid ist eine elastisch-harte Raumforderung tastbar.
Erstsymptom: In etwa der Hälfte der Fälle eine schmerzlose einseitige Augapfelverlagerung, in der anderen Hälfte eine Ptosis.
Adenoid-zystisches Karzinom
Schnelles Wachstum : schnelleres Wachstum als pleomorphes Adenom. Die Symptomdauer beträgt oft weniger als 6 Monate.
Schmerzen : häufige Schmerzen aufgrund von Nerveninfiltration.
Perineurale Infiltration : bis zu 85 % der Fälle. Kann zu Hypästhesie in den Versorgungsgebieten des ersten und zweiten Astes des Trigeminusnervs führen.
Plötzliche Schmerzzunahme : wichtiges Zeichen, das auf eine maligne Transformation hindeutet.
Lymphom der Tränendrüse
Bevorzugtes Alter : tritt häufig nach dem 60. Lebensjahr auf.
Bilateralität : etwa 25 % der Fälle sind bilateral.
Systemische Beteiligung : etwa 34 % der Fälle sind mit einem systemischen Lymphom assoziiert.
MALT-Typ : langsamer, schmerzloser Verlauf. Der großzellige Typ zeigt eine schnelle Progression und Entzündungszeichen.
S-förmige Deformität (Deformität des Oberlids) und Abweichung des Augapfels nach unten oder unten-innen sind häufige Befunde bei Tränendrüsentumoren. Ein Entropium des Unterlids kann ebenfalls auftreten.
Im von Zhong et al. berichteten Fall eines 60-jährigen Mannes mit MALT-Lymphom der Tränendrüse betrug der Exophthalmus rechts 17 mm und links 12 mm mit einer inferonasalen Abweichung. Die CT zeigte eine 3,3 × 2,3 × 2,4 cm große Raumforderung in der rechten Tränendrüse. 1)
QWas ist der Unterschied zwischen den Symptomen des pleomorphen Adenoms und des adenoid-zystischen Karzinoms?
A
Das pleomorphe Adenom ist durch ein langsames, schmerzloses Wachstum gekennzeichnet, mit einer durchschnittlichen Symptomdauer von etwa 2 Jahren. Das adenoid-zystische Karzinom hingegen wächst schneller als das pleomorphe Adenom und ist häufiger schmerzhaft. Da eine perineurale Infiltration in bis zu 85 % der Fälle auftritt, können auch sensorische Anomalien im Gesicht ein Hinweis sein. Die endgültige Unterscheidung zwischen gutartig und bösartig ist jedoch nur durch eine histopathologische Untersuchung möglich.
Alter: Pleomorphes Adenom tritt häufig im Alter von 30–40 Jahren auf, adenoidzystisches Karzinom von der Jugend bis ins hohe Alter, Lymphom nach dem 60. Lebensjahr.
Lymphom in der Vorgeschichte: Infiltration der Tränendrüse durch ein systemisches Lymphom.
Zustand nach unvollständiger Resektion: Erhöht das Risiko eines Rezidivs und einer malignen Transformation des pleomorphen Adenoms.
Rezidiv eines pleomorphen Adenoms nach dem 45. Lebensjahr: Wiederholte Rezidive erhöhen das Risiko einer malignen Transformation.
Adenoidzystisches Karzinom: Gekennzeichnet durch das MYB-NFIB-Fusionsgen. Beteiligt sind Translokationen an 6q22-23 und 9pq23-24.
Maligne Transformation des pleomorphen Adenoms: Es wurden Fälle von Transformation in ein Karzinom ex pleomorphem Adenom 5–20 Jahre nach der Exzision berichtet.
Das MALT-Lymphom entsteht durch chronische antigene Stimulation, die den NF-κB-Signalweg konstitutiv aktiviert und zur tumorösen Proliferation von B-Zellen führt. 1)
Die Helicobacter pylori-Infektion beim gastralen MALT-Lymphom ist ein klassisches Modell für den Übergang von Entzündung zur Tumorentstehung. 1)
Beim MALT-Lymphom der Tränendrüse wurde in Italien und Korea in über 50 % der Fälle ein Zusammenhang mit Chlamydia psittaci berichtet. 1)
Auch Autoimmunerkrankungen (rheumatoide Arthritis, Hashimoto-Thyreoiditis, Sjögren-Syndrom) erhöhen das Risiko für MALT-Lymphome. 1)
Anamnese: Überprüfung des Beginns, des Verlaufs der Verschlechterung, des Vorhandenseins von Schmerzen, der Operationsgeschichte und anderer Erkrankungen (Autoimmunerkrankungen, bösartige Tumore usw.).
Palpation: Beurteilung von Größe, Konsistenz, Verwachsungen und Druckschmerz der Raumforderung. Auch die Schwellung der regionalen Lymphknoten wird überprüft.
Die folgende Tabelle zeigt die Merkmale der wichtigsten bildgebenden Untersuchungen.
Untersuchung
Bewertungsgegenstand
Bildgebende Merkmale des pleomorphen Adenoms
CT
Knochenveränderungen, Verkalkungen
Scharfe Begrenzung, kugelförmig bis oval
MRT
Weichteile
T1 hypo- bis isointens / T2 hyperintens, moderate Kontrastmittelaufnahme
PET/CT
Ganzkörper-Metastasenbeurteilung
—
CT : Nützlich zur Beurteilung von Knochenveränderungen und Verkalkungen. Beim pleomorphen Adenom zeigen sich eine kompressionsbedingte Erweiterung der Tränendrüsengrube, Knochensclerose und Knochenverdünnung. Maligne Tumoren sind oft unscharf begrenzt. Tumorgröße, -lage und Vorhandensein von Knochendefekten sind für die Operationsplanung unerlässlich.
MRT : Hervorragend zur Beurteilung von Weichteilen. Das pleomorphe Adenom zeigt in T1-gewichteten Aufnahmen eine Signalintensität, die der der äußeren Augenmuskeln entspricht oder niedriger ist, in T2-gewichteten Aufnahmen eine höhere Signalintensität und eine moderate Kontrastmittelaufnahme. Große Tumoren können innere zystische Degeneration oder Verkalkungen aufweisen. Die MRT gilt als Goldstandard für die Diagnose von okulären Adnexlymphomen. 1)
PET/CT und Galliumszintigraphie : Werden zur Beurteilung eines systemischen Lymphoms oder von Fernmetastasen eingesetzt.
Es ist oft schwierig, ein pleomorphes Adenom allein anhand bildgebender Befunde von einem malignen epithelialen Tumor zu unterscheiden; eine Gesamtbeurteilung in Kombination mit klinischen Informationen ist erforderlich.
Die Biopsiestrategie variiert je nach vermuteter Erkrankung.
Verdacht auf Lymphom oder bösartigen Tumor : Durchführung einer perkutanen Exzisionsbiopsie. Über einen lateralen Schnitt unterhalb der Augenbraue wird mindestens etwa 5 mm³ Tumor entfernt.
Verdacht auf pleomorphes Adenom : Nadelbiopsie oder Exzisionsbiopsie sind grundsätzlich zu vermeiden, da das Risiko einer Tumoraussaat und eines Rezidivs durch Kapselruptur besteht.
Durchflusszytometrie und Gen-Rearrangement-Tests : Werden bei Verdacht auf Lymphom durchgeführt, um Monoklonalität zu bestätigen.
Tränendrüsenzyste: Durch Diaphanoskopie erkennbar.
Dakryoadenitis: viral oder autoimmun. Etwa 30 % der Tränendrüsenbiopsien werden als idiopathische Dakryoadenitis diagnostiziert.
IgG4-assoziierte Augenerkrankung (Mikulicz-Krankheit): Charakteristisch ist eine beidseitige Drüsenschwellung. Die Bestimmung des IgG4-Spiegels ist hilfreich.
Reaktive lymphatische Hyperplasie: Zur Abgrenzung von einem malignen Lymphom ist eine Pathologie unerlässlich.
QIst eine Biopsie auch bei Verdacht auf ein pleomorphes Adenom erforderlich?
A
Bei Verdacht auf ein pleomorphes Adenom ist es grundsätzlich zu vermeiden, eine Biopsie durchzuführen. Denn wenn die Kapsel während der Biopsie reißt, werden Tumorzellen im umliegenden Gewebe ausgesät, was die Rezidivrate erheblich erhöht. Wenn die Bildgebung stark auf ein pleomorphes Adenom hindeutet, wird direkt eine vollständige En-bloc-Resektion durchgeführt. Wenn hingegen ein Lymphom oder eine entzündliche Erkrankung vermutet wird, ist eine Probeexzision indiziert.
Bei einem pleomorphen Adenom ist eine frühzeitige Operation nach der Entdeckung wünschenswert. Denn wenn der Tumor zu groß wird, wird die Operation schwierig.
En-bloc-Resektion ist das Prinzip: Der Tumor muss bei der ersten Operation ohne Kapselruptur en bloc entfernt werden. Bei unvollständiger Resektion kommt es zu wiederholten Rezidiven.
Nadelbiopsie und Probeexzision sind kontraindiziert: Sie führen zu Tumoraussaat und Rezidiven.
Pleomorphes Adenom des Augenlids: Kann unter Lokalanästhesie über einen verlängerten Schnitt in der Lidfalte entfernt werden.
Pleomorphes Adenom der Orbita: Eine Osteotomie (knochenbildende Knochenresektion) ist erforderlich. Ein pleomorphes Adenom in der Tränendrüsengrube wird bei der Entfernung oft durch den Knochen des Orbitarands behindert, und eine vollständige Resektion ohne Osteotomie ist schwierig. Das chirurgische Vorgehen umfasst eine Osteotomie von der Incisura supraorbitalis bis zum oberen Rand des Jochbogens, die Entfernung des Tumors, das Zurücksetzen des Knochens und die Fixierung durch Periostnaht.
Problem der unvollständigen Resektion: Bei unvollständiger Resektion steigt das Rezidivrisiko erheblich, und wiederholte Rezidive erhöhen das Risiko einer malignen Transformation.
Nicht vollständig resezierbare Fälle: Nach Probeexzision weite Exzision (Orbitaexenteration) + Strahlentherapie. Auch eine externe Bestrahlung nach Tumordebulking ist eine Option.
Neoadjuvante Chemotherapie: Eine neoadjuvante Chemotherapie über die A. carotis externa wird in einigen Fällen durchgeführt.
Schwerionentherapie: Befindet sich in der klinischen Erprobung und wird als Option mit Potenzial zum Orbitaerhalt erforscht (siehe Abschnitt « Aktuelle Forschung »).
Die Prognose variiert stark je nach histologischem Typ.
Pleomorphes Adenom: Da es sich um einen gutartigen Tumor handelt, ist die Prognose gut, aber eine langfristige Nachsorge ist erforderlich. 5–20 Jahre nach der Entfernung kann eine maligne Transformation zum Karzinom ex pleomorphem Adenom erfolgen. Bei inkompletter Resektion besteht ein hohes Rezidivrisiko.
Adenoidzystisches Karzinom: Mittlere Überlebenszeit 36 Monate, 10-Jahres-Überlebensrate 20–30 %, ungünstige Prognose. Etwa 50 % entwickeln Fernmetastasen (häufig Lunge und Knochen). Starke Nerven- und Lymphgefäßinvasion, möglich ist auch eine Invasion des Hirnstamms. Hochmaligner Verlauf mit Knochenzerstörung.
Primäres Adenokarzinom: 3-Jahres-Mortalität 70 %, extrem ungünstige Prognose.
Mukosaassoziiertes lymphatisches Gewebe (MALT)-Lymphom: Wenig Metastasen, gute Ansprechrate auf Strahlentherapie, relativ gute Prognose.
Diffuses großzelliges B-Zell-Lymphom: Hohe Metastasierungsneigung und ungünstige Prognose.
QWie wird das maligne Lymphom (MALT-Typ) behandelt?
A
Bei lokalisiertem MALT-Lymphom sind Überwachung oder Strahlentherapie (30 Gy) die Erstlinientherapie. Bei CD20-Positivität ist Rituximab (375 mg/m² wöchentlich für 4 Zyklen) wirksam. Bei systemischer Metastasierung ist eine Chemotherapie wie R-CHOP indiziert. Das MALT-Lymphom spricht gut auf Strahlentherapie an und hat eine relativ gute Prognose.
Es handelt sich um einen gut abgegrenzten Tumor, der von einer Pseudokapsel umgeben ist. Charakteristisch ist ein biphasisches Muster mit einer Mischung aus epithelialen und myoepithelialen Zellen sowie mesenchymalen Komponenten, was den Namen „pleomorph“ erklärt.
Die Tumorzellen proliferieren unter Bildung von Lumina. Die Lumina-Wand ist zweischichtig: Die äußere Schicht besteht aus kleinen kubischen oder spindelförmigen Zellen mit myoepithelialem Charakter. Diese myoepithelialen Zellen setzen sich ins Stroma fort und produzieren durch Metaplasie Schleim (mit Glykosaminoglykanen) und mesodermale Substanzen wie Knorpel.
Äußere Schicht (myoepitheliale Zellen) : produziert muzinöse und chondroide Matrix.
Innere Schicht (epitheliale Zellen) : bildet gangartige Strukturen, die Glykoproteine sezernieren.
Die Pseudokapsel ist dünn und unvollständig, was bei Kapselruptur leicht zu einer Tumoraussaat führt.
Charakteristisch sind solide und trabekuläre Wachstumsmuster sowie eine ausgeprägte perineurale Invasion. Die Tumorzellen sind klein mit chromatinreichen Kernen. Die perineurale Invasion kann durch Neurofilamentfärbung nachgewiesen werden.
Die histologischen Subtypen werden in fünf Kategorien eingeteilt.
Kribriformer Typ : der häufigste Subtyp. Gekennzeichnet durch siebförmige Hohlraumbildung.
Solider Typ
Sklerosierender Typ
Komedokarzinomatöser Typ
Tubulärer Typ
Polymorphes Adenokarzinom und Mukoepidermoidkarzinom
Polymorphes Adenokarzinom : maligne Transformation innerhalb eines pleomorphen Adenoms. Wenige Drüsenstrukturen und geringer Differenzierungsgrad.
Mukoepidermoidkarzinom : Mischung aus Schleimzellen, epidermoiden Zellen, Intermediärzellen, Zylinderzellen und hellen Zellen, mit zystischen Komponenten.
Diffuse Proliferation lymphozytenähnlicher Zellen, wobei eine monoklonale B-Zell-Proliferation durch Immunhistochemie und Southern Blot nachgewiesen werden kann.
In dem von Zhong et al. berichteten Fall zeigte die immunhistochemische Färbung CD20-positiv, CD79a-positiv, PAX5-positiv, CD10-positiv, BCL2-negativ, BCL6-positiv. Der Ki67-Proliferationsindex betrug etwa 60% im Keimzentrum und etwa 15% im plasmazytoiden Bereich. 1)
MYB-NFIB-Fusionsgen : Die Fusion des Transkriptionsfaktors MYB mit NFIB führt zur Überexpression der Zielgene von MYB, was zu Anomalien in Zellproliferation, -überleben und -differenzierung führt.
Chromosomentranslokation : Beteiligung der Regionen 6q22-23 und 9pq23-24.
Molekulare Mechanismen des Mukosa-assoziierten lymphatischen Gewebe-Lymphoms
Chronische Antigenstimulation (Infektion mit Chlamydia psittaci, Autoimmunreaktion) aktiviert dauerhaft das B-Zell-Rezeptor-Signal. 1)
Dies führt zur konstitutiven Aktivierung des NF-κB-Signalwegs, der die Proliferation und das Überleben von Tumor-B-Zellen fördert. 1)
Das Helicobacter pylori-assoziierte gastrische MALT-Lymphom ist ein klassisches Modell, bei dem die Eradikation der Infektion zur Tumorrückbildung führt. 1)
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Die Schwerionentherapie (Kohlenstoffionen) wird derzeit in klinischen Studien hauptsächlich für das adenoidzystische Karzinom untersucht. Im Vergleich zur herkömmlichen externen Strahlentherapie bietet der Bragg-Peak eine höhere Dosiskonzentration, wodurch eine Hochdosisbestrahlung unter Schonung der Augenhöhle möglich ist. Derzeit befindet sie sich in der klinischen Erprobung und ist nicht als Standardbehandlung etabliert.
Die Fluoreszenz-in-situ-Hybridisierung (FISH) gilt als wichtiges Mittel zur molekulargenetischen Bestätigung bei der Diagnose von Lymphomen des mukosaassoziierten lymphatischen Gewebes. 1)
Durch die Überwachung der Immunglobulin-Schwerketten- (IGH) und Immunglobulin-Kappa-Ketten- (IGK) Genumlagerungen wird erwartet, dass die Beurteilung des Behandlungserfolgs und der Nachweis einer minimalen Resterkrankung möglich werden. 1)
Es wurde vermutet, dass das Verhältnis von regulatorischen T-Zellen (Treg) zu T-Helfer-17-Zellen mit dem Tumorausgang zusammenhängen könnte, und die Aufklärung des Immunmikromilieus soll zur Suche nach neuen therapeutischen Zielstrukturen führen. 1)
Zhong Q, Yan Y, Li S.. Mucosa-associated lymphoid tissue lymphoma of the lacrimal gland: A case report and literature review. Medicine (Baltimore). 2024;103(21):e38303. doi:10.1097/md.0000000000038303. PMID:38787969; PMCID:PMC11124633.