Kribriformer Typ
Häufigkeit: am häufigsten (39,8 %)
Merkmale: Läppchenstrukturen mit runden Muzinansammlungen. „Schweizerkäse“-Aussehen. Mittlere Prognose.

Das adenoidzystische Karzinom (ACC) ist ein seltener bösartiger Tumor der Sekretdrüsen mit einer weltweiten Inzidenz von 3–4 Fällen pro Million Einwohner 1). Es macht etwa 1 % aller Kopf-Hals-Karzinome aus, und wenn es in der Tränendrüse auftritt, spricht man vom adenoidzystischen Karzinom der Tränendrüse 1).
Das adenoidzystische Karzinom der Tränendrüse macht etwa 1,6 % aller Orbitatumoren aus 3)5) und ist der häufigste maligne epitheliale Tumor der Tränendrüse, der etwa 13–40 % der Tränendrüsenkarzinome ausmacht 5). Tränendrüsentumoren machen etwa 10 % der raumfordernden Orbitaprozesse aus, etwa 20 % der soliden Tränendrüsentumoren sind epithelialen Ursprungs, davon sind etwa 45 % bösartig, und etwa 60 % der malignen epithelialen Tränendrüsentumoren sind adenoidzystische Karzinome.
Das adenoidzystische Karzinom der Tränendrüse wurde erstmals von Theodor Billroth beschrieben und aufgrund seiner histologischen Merkmale ursprünglich als „Zylindrom“ bezeichnet 5).
Die epidemiologischen Merkmale sind wie folgt.
Es macht etwa 1,6 % aller orbitalen Tumoren aus, und die weltweite Inzidenz des adenoid-zystischen Karzinoms liegt bei 3–4 Fällen pro Million Einwohner, was extrem selten ist1)5). Es ist der häufigste histologische Typ unter den malignen epithelialen Tumoren der Tränendrüse.
Laut einer großen Übersicht von 806 Fällen ist die Häufigkeit der Erstsymptome wie folgt5).
| Symptom | Häufigkeit |
|---|---|
| Exophthalmus | 27,4 % |
| Schmerz | 21,7 % |
| Lidschwellung | 10,9 % |
| Augapfeldeviation | 10,2 % |
| Eingeschränkte Augenbeweglichkeit | 10,1 % |
| Sehverschlechterung | 9,3 % |
| Doppelbilder | 6,7 % |
| Ptosis | 4,1 % |
Schmerz ist ein charakteristisches Symptom des adenoid-zystischen Karzinoms, verursacht durch perineurale Infiltration (PNI). Es ist ein wichtiges Unterscheidungsmerkmal zu gutartigen Tumoren. Der Augapfel ist häufig nach innen unten abgewichen, da die Tränendrüse im oberen äußeren Bereich der Orbita liegt. Es können auch frontotemporale Hypästhesie oder S-förmige Ptosis auftreten. Die durchschnittliche Zeit vom Symptombeginn bis zur Diagnose beträgt 11,1 ± 18,3 Monate (Spanne 0,5 bis 120 Monate) 5).
Die Wachstumsgeschwindigkeit des Tumors ist schneller als die des pleomorphen Adenoms, und er neigt zu Schmerzen, was ein wichtiger Punkt für die Differentialdiagnose ist. Exophthalmus durch Tumorvergrößerung, Bewegungseinschränkung der Augen durch Infiltration der äußeren Augenmuskeln und Nerven sowie Sehverschlechterung durch Kompression des Sehnervs schreiten ebenfalls relativ schnell voran.
Das Vorhandensein oder Fehlen von Schmerzen ist der wichtigste Unterscheidungspunkt. Das adenoid-zystische Karzinom verursacht durch perineurale Infiltration Schmerzen, während gutartige Tumoren (wie das pleomorphe Adenom) schmerzlos sind und langsam fortschreiten. Wenn im CT eine Knochenzerstörung nachweisbar ist, kann man von einem bösartigen Tumor ausgehen, aber es gibt auch adenoid-zystische Karzinome ohne Knochenzerstörung; bei Schmerzen sollte daher aktiv eine Biopsie in Betracht gezogen werden.
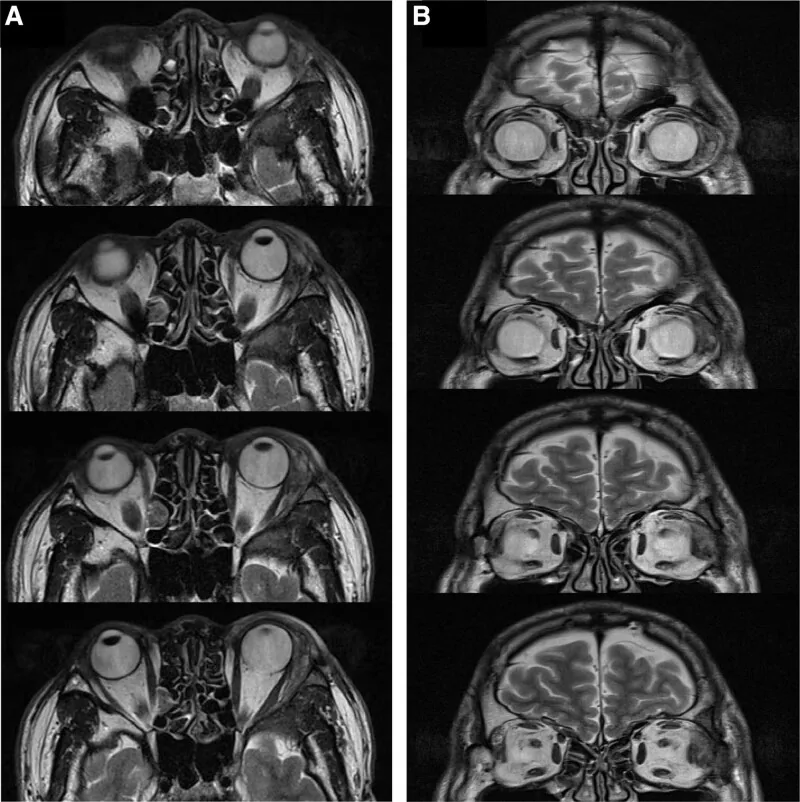
Laut dem Bericht von Williams et al. wurde histologisch bei 82 % der Patienten eine Infiltration der Tränendrüsengrube bestätigt1). Wenn im CT der Tumor den Orbitaknochen zerstört, kann man von einem bösartigen Tumor ausgehen, aber es ist zu beachten, dass es auch adenoid-zystische Karzinome ohne Knochenzerstörung gibt.
Die Ursache des adenoid-zystischen Karzinoms ist unbekannt, und es sind keine spezifischen Risikofaktoren etabliert. Der Tumor entsteht am häufigsten aus dem Orbitallappen der Tränendrüse und ist ein nicht gekapselter Tumor.
Die folgenden pathologischen und klinischen Merkmale sind als Faktoren für eine schlechte Prognose bekannt:
Die definitive Diagnose basiert auf der histopathologischen Beurteilung. Bei Verdacht auf Malignität wird eine Exzisionsbiopsie empfohlen, um eine intraorbitale Aussaat von Tumorzellen zu vermeiden. Eine Feinnadelaspirationsbiopsie kann bei nicht resezierbaren Tumoren geeignet sein, jedoch nur in Einrichtungen mit erfahrenem Zytopathologen.
Bei Symptomen wie Exophthalmus, die auf einen Tränendrüsentumor hindeuten, werden ophthalmologische Untersuchungen (Sehschärfe, Gesichtsfeld, Augenbeweglichkeit) sowie eine Bildgebung mittels nativer CT und nativer/kontrastverstärkter MRT durchgeführt. Falls eine systemische Suche erforderlich ist, können PET-CT oder Kontrast-CT erfolgen. Die definitive Diagnose erfolgt durch Biopsie oder histopathologische Untersuchung nach vollständiger Resektion.
| T-Stadium | Definition |
|---|---|
| T1 | Maximaler Durchmesser ≤ 2 cm |
| T2 | Maximaler Durchmesser > 2 cm bis ≤ 4 cm |
| T3 | Maximaldurchmesser > 4 cm oder Ausdehnung in orbitales Weichgewebe |
| T4 | Infiltration der Nasennebenhöhlen, Fossa temporalis, Fossa pterygopalatina, Fissura orbitalis superior, Sinus cavernosus oder des Gehirns |
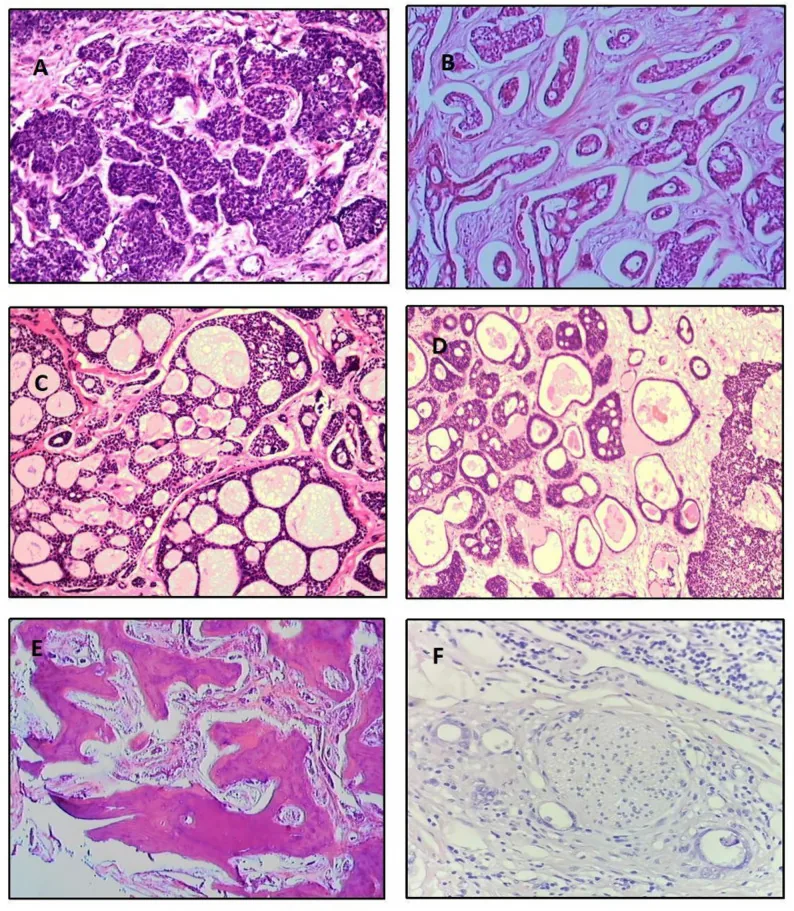
Die Häufigkeit der histologischen Subtypen basierend auf einer Analyse von 515 Fällen ist wie folgt 5).
Kribriformer Typ
Häufigkeit: am häufigsten (39,8 %)
Merkmale: Läppchenstrukturen mit runden Muzinansammlungen. „Schweizerkäse“-Aussehen. Mittlere Prognose.
Basaloider Typ
Häufigkeit: 31,8 %
Merkmale: schlecht differenziert. Große basophile Kerne und spärliches Zytoplasma. Schlechteste Prognose.
Tubulärer Typ
Häufigkeit: 7,4 %
Merkmale: Epithelschläuche, ausgekleidet mit 2–3 Zellschichten. Am höchsten differenziert, beste Prognose.
Daneben gibt es gemischte (13,9 %), undifferenzierte (6,1 %) und sklerosierende (0,9 %) Typen. Ein solides Muster >30 % gilt als ungünstige Prognose1). Histologisch sind die Tumorzellen klein, das Zytoplasma spärlich und bläulich, der Kern chromatinreich. Die Grenze zwischen Tumorzellnestern und Stroma ist scharf, deutlich unterschiedlich vom pleomorphen Adenom.
Die Operation ist die Grundlage der Behandlung, und das Verfahren wird durch das Tumorstadium und die Bildgebung bestimmt3)5).
Die Kaplan-Meier-Analyse zeigte, dass die augenerhaltende Operation + Strahlentherapie eine bessere Überlebensrate aufwies als die Exenteration ± Strahlentherapie (P < 0,05)5).
Wenn die Bildgebung einen kleinen, vollständig resezierbaren Tumor zeigt, wird eine vollständige Resektion angestrebt. Ist eine vollständige Resektion nicht möglich, wird eine Probebiopsie zur pathologischen Bestätigung durchgeführt, dann eine weite Exzision oder Strahlentherapie erwogen. Beim adenoid-zystischen Karzinom der Tränendrüse, das auf die Orbita beschränkt ist, kann eine Exenteration in Betracht gezogen werden, aber unter Berücksichtigung kosmetischer Probleme und des Alters/Wunsches des Patienten kann auch eine konservative Behandlung gewählt werden. Lymphknotenmetastasen sind selten (4–9 %), eine Lymphknotendissektion ist in der Regel nicht erforderlich6).
Selbst nach einer weiten Resektion und Strahlentherapie kann eine Infiltration des Hirnstamms über den Tränennerv manchmal nicht verhindert werden. Auch Fernmetastasen können nicht immer vermieden werden, und eine langfristige Nachsorge ist erforderlich.
Für inoperable adenoid-zystische Karzinome wird eine Schwerionen-Strahlentherapie durchgeführt, die als vielversprechende Behandlung gilt, um den Tumor zu kontrollieren und gleichzeitig Augenlid, Augapfel und Orbita zu erhalten.
NIAC wurde erstmals 1998 von Meldrum et al. berichtet, wobei eine Kombination aus intraarteriellem Cisplatin (100 mg/m²) und intravenösem Doxorubicin verwendet wird 3)2).
In einer Studie von Tse et al. (2013) mit 19 Fällen wurde bei 8 Fällen, bei denen die Tränenarterie erhalten und das Protokoll eingehalten wurde, ein 10-Jahres-krankheitsfreies Überleben von 100 % berichtet 2)3).
Die Kombination aus NIAC + Resektion/Exenteration + Strahlentherapie zeigt im Vergleich zu anderen Behandlungen gute Ergebnisse mit einer Rezidivrate von 10,8 %, einer Metastasierungsrate von 14,9 % und einer Mortalitätsrate von 18,9 % 5). Zu den Hauptrisiken von NIAC gehören vorübergehende Gesichtslähmung, Sehverlust, anteriore Ischämie, Neutropenie und Thrombozytopenie 3).
Die Kaplan-Meier-Analyse zeigte, dass das Überleben nach augenerhaltender Operation plus Strahlentherapie besser war als nach Orbitaeviszeration ± Strahlentherapie (P<0,05)5). Für T1-T2-Tumoren wird der Augenerhalt empfohlen, für T3-T4-Tumoren oder bei extraorbitaler Ausdehnung wird eine Orbitaeviszeration in Betracht gezogen. Aufgrund kosmetischer Probleme oder Patientenwunsch kann jedoch auch eine konservative Behandlung gewählt werden.
Die Tumorzellen sind klein, mit spärlichem, bläulichem Zytoplasma und chromatinreichen Kernen. Histologisch ist die Grenze zwischen Tumorzellnestern und Stroma klar, deutlich unterschieden vom pleomorphen Adenom.
Beim kribriformen Typ sind echte Lumina (von Gangzellen abstammend) und Pseudolumina (von Myoepithelzellen gebildete Muzinhohlräume) gemischt, was ein „Schweizer Käse“-Aussehen ergibt. Der sklerosierende Typ zeigt sich als Epithelzellstränge mit dichtem hyalinisiertem Stroma. Ein solides Muster von über 30 % gilt als prognostisch ungünstig1).
Die Wachstumsmuster des Tumors werden in fünf Typen eingeteilt (kribriform, solide, sklerosierend, komedokarzinomatös, tubulär). In derselben Probe können mehrere Wachstumsmuster beobachtet werden.
| Molekulare Anomalie | Inhalt |
|---|---|
| MYB-NFIB-Fusion | Translokation t(6;9)(q23;p23). Bei über 70% aller adenoid-zystischen Karzinome vorhanden1) |
| Überexpression von MYB | Fördert Zellproliferation, Differenzierung, Angiogenese und Hochregulation von Wachstumsfaktoren1) |
| Aktivierende NOTCH1-Mutation | Haupttreiber für Proliferation und Invasion bei metastasiertem adenoid-zystischem Karzinom1)2) |
| KRAS/NRAS/MET-Mutationen | Berichtet in 46%, 8% bzw. 13% der Fälle. EGFR-RAS-RAF-Kaskade als mögliches therapeutisches Ziel1)5) |
Beim adenoid-zystischen Karzinom der Tränendrüse wird in 58% der Fälle eine MYB-Umlagerung nachgewiesen (Mayo Clinic, 12 Fälle/25 Jahre), und die MYB-NFIB-Fusion ist ein hochspezifischer diagnostischer Marker für das adenoid-zystische Karzinom1). Die MYB-NFIB-Fusion wird durch AKT-abhängige IGF1R-Signalgebung reguliert, und die IGF1R-Hemmung gilt als vielversprechendes therapeutisches Ziel1)3).
Eine perineurale Infiltration wird in 45,3% der Fälle beobachtet, und Tumorzellen können sich auch ohne vaskuläre oder lymphatische Infiltration ausbreiten1). Es besteht eine starke Tendenz zur Infiltration von Nerven und Lymphgefäßen, und eine Infiltration des Hirnstamms über den Tränennerv kann bereits im Frühstadium der Erkrankung auftreten. Die perineurale Infiltration wird mit einer Apoptoseresistenz durch Hochregulation von Bcl-2 in Verbindung gebracht.
Derzeit ist kein Medikament der molekularen Zielgerichteten Therapie für das adenoid-zystische Karzinom der Tränendrüse zugelassen. Folgende Ziele werden in der Forschung untersucht 1)3).
Die Ergebnisse klinischer Phase-II-Studien berichten: Dovitinib (ORR 6%, mPFS 8,2 Monate), Lenvatinib (ORR 16%, mPFS 17,5 Monate), Axitinib (ORR 9%, mPFS 5,7 Monate) 3).
Yu et al. (2022) berichteten über eine Bewertung, die Genomsequenzierung vor und nach der Behandlung mit der Analyse von Apoptose-Markern (cCas3, PARP) kombinierte 2). Die Mutationsallelfrequenz (VAF) der NOTCH1-Mutation nach NIAC sank von 18,07 % vor der Behandlung auf 11,34 % nach der Behandlung (37 % Reduktion), was auf einen möglichen prädiktiven Marker für die Cisplatin-Empfindlichkeit hindeutet.
Derzeit gibt es kein zugelassenes zielgerichtetes Medikament für das adenoid-zystische Karzinom der Tränendrüse. MYB-NFIB-Fusion, Notch-Signalweg, EGFR-RAS-RAF-Kaskade usw. werden als potenzielle therapeutische Ziele untersucht, und mehrere Phase-II-Studien laufen1)3). Für Patienten, die eine Standardbehandlung wünschen, ist die Kombination aus Operation und Strahlentherapie derzeit die Option.