
IgG4-assoziierte Augenerkrankung (Mikulicz-Krankheit usw.)
1. Was ist die IgG4-assoziierte Augenerkrankung?
Abschnitt betitelt „1. Was ist die IgG4-assoziierte Augenerkrankung?“Die IgG4-assoziierte Augenerkrankung (IgG4-related ophthalmic disease; IgG4-ROD) ist eine Erkrankung unbekannter Ursache, bei der das Serum-IgG4 erhöht ist und IgG4-positive Plasmazellen in die Orbita infiltrieren, was zu Tumorbildung und Gewebeverdickung führt. Sie wird als orbitale Lokalisation der systemischen IgG4-assoziierten Erkrankung (IgG4-RD) betrachtet.
Die IgG4-RD ist eine Erkrankung, bei der Plasmazellen, die IgG4, eines der Serum-Immunglobuline, produzieren, in verschiedene Organe des Körpers infiltrieren und so zur Bildung von Tumoren und Gewebeverdickung führen. Das Konzept wurde 2001 als Ursache der Autoimmunpankreatitis etabliert. Im ophthalmologischen Bereich wurde 2004 erstmals über die IgG4-assoziierte Mikulicz-Krankheit berichtet, und später verbreitete sich das Konzept der IgG4-assoziierten Erkrankung in der Augenheilkunde. Die Diagnosekriterien wurden erstmals 2014 festgelegt 1) und 2023 überarbeitet 1).
In Japan sind die häufigsten primären Orbitatumoren lymphoproliferative Erkrankungen, zu denen malignes Lymphom, reaktive lymphatische Hyperplasie, IgG4-ROD und idiopathische orbitale Entzündung (IOI) gehören. Allein lymphoproliferative Erkrankungen machen 50–60 % aller Orbitatumoren aus. Es hat sich gezeigt, dass 17–60 % der Fälle, die als idiopathische orbitale Entzündung diagnostiziert wurden, tatsächlich eine IgG4-ROD waren. Viele Fälle der früheren Mikulicz-Krankheit und der spezifischen orbitalen Entzündung wurden in diese Erkrankung umklassifiziert.
Die IgG4-ROD tritt bei Männern und Frauen gleichermaßen auf, das Durchschnittsalter liegt bei etwa 55–60 Jahren, und Fälle unter 20 Jahren sind selten.
Lokalisation der IgG4-ROD
Abschnitt betitelt „Lokalisation der IgG4-ROD“In einer multizentrischen Studie mit 378 Fällen war die Verteilung der Lokalisationen wie folgt 1):
| Lokalisation | Häufigkeit |
|---|---|
| Tränendrüse | 62–88 % (86 % in der Studie mit 378 Fällen) |
| Orbitalfett | 28,6–40 % |
| Äußere Augenmuskeln | 19–25 % (21 % in der Studie mit 378 Fällen) |
| Perineural des Trigeminus | 9,5–39 % (20 % in einer Studie mit 378 Fällen) |
| Augenlid | 12 % |
| Tränen-Nasen-Gang-System | 1,5–9,5 % |
In derselben Studie betrug die Verteilung der subjektiven Symptome: trockenes Auge 22 %, Doppelbilder 20 %, Sehverschlechterung 8 %, Gesichtsfeldausfall 5 %1).
Das normale Serum-IgG4 liegt unter 135 mg/dL und ist durch drei Hauptläsionen gekennzeichnet (Tränendrüsenschwellung, perineurale Raumforderung des Trigeminus, Schwellung der äußeren Augenmuskeln). Weitere Orbitalläsionen umfassen Schwellung des orbitalen Fettgewebes, Raumforderungen um den Sehnerv und Blutgefäße, sowie Läsionen der Lidhaut, Bindehaut, Tränensack und Sklera.
2. Hauptsymptome und klinische Befunde
Abschnitt betitelt „2. Hauptsymptome und klinische Befunde“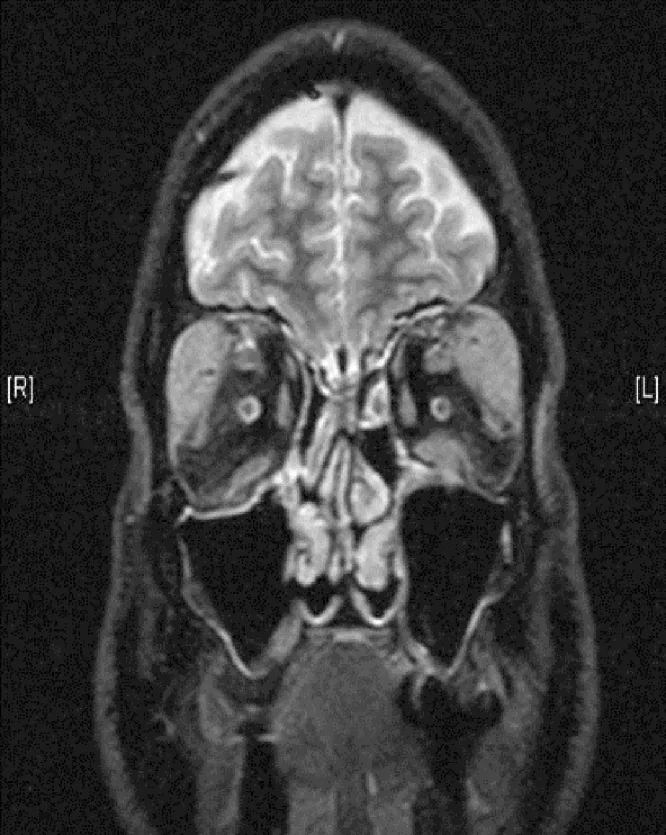
Subjektive Symptome
Abschnitt betitelt „Subjektive Symptome“- Lidschwellung: durch Tränendrüsenvergrößerung. Typischerweise schmerzlos und langsam fortschreitend. Oft symmetrisch, und wenn sie mit einer Speicheldrüsenschwellung einhergeht, wird sie als Mikulicz-Krankheit klassifiziert.
- Exophthalmus: durch Schwellung der äußeren Augenmuskeln und des orbitalen Weichgewebes.
- Doppelbilder: kann aufgrund einer Beteiligung der äußeren Augenmuskeln zu einem restriktiven Strabismus führen. Der Musculus rectus inferior ist am häufigsten betroffen.
- Verschlechterung des Sehvermögens und Gesichtsfeldausfälle: verursacht durch kompressive Optikusneuropathie. In einer Studie mit 378 Fällen wurde bei 8 % eine Sehverschlechterung und bei 5 % Gesichtsfeldausfälle festgestellt1).
- Trockenes Auge: trat in einer Studie mit 378 Fällen bei 22 % auf. Da die Zerstörung des Tränendrüsengewebes jedoch begrenzt ist, ist es oft weniger ausgeprägt als beim Sjögren-Syndrom.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Abschnitt betitelt „Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)“- Vergrößerung der Tränendrüsen: Lidödem mit S-förmiger Deformität. In der MRT ist die bilaterale Tränendrüsenvergrößerung am häufigsten.
- Vergrößerung des Nervus trigeminus: Raumforderungen um den Nervus supraorbitalis und infraorbitalis. Dies ist ein charakteristischer Befund der IgG4-ROD und ein Unterscheidungsmerkmal zur endokrinen Orbitopathie.
- Vergrößerung der äußeren Augenmuskeln: Abgrenzung zur endokrinen Orbitopathie erforderlich.
- Perineurale Läsionen des Sehnervs: können eine Optikusneuropathie verursachen. Bei hohen IgG4-Werten über 500 mg/dL ist besondere Vorsicht geboten.
- MRT-Befunde: isointens in T1-gewichteten Aufnahmen, hypointens in T2-gewichteten Aufnahmen, homogene Kontrastmittelaufnahme mit Gadolinium.
Systemische Läsionen treten bei 68 % der Patienten mit IgG4-ROD auf. Die häufigsten Begleitlokalisationen sind Speicheldrüsen (43 %), Lymphknoten (27 %) und Bauchspeicheldrüse (20 %).
Beide können eine Vergrößerung der äußeren Augenmuskeln und einen Exophthalmus verursachen. Zur Unterscheidung sind der Serum-IgG4-Spiegel, Schilddrüsenfunktionstests (T3, T4, TSH), MRT-Befunde (bei IgG4-ROD T2-Hypointensität, häufige Trigeminus-Hypertrophie) und eine Gewebebiopsie wichtig. Die IgG4-ROD geht häufig mit einer Tränendrüsenvergrößerung einher, während bei der endokrinen Orbitopathie die Vergrößerung des Musculus rectus inferior und medialis überwiegt.
3. Ursachen und Risikofaktoren
Abschnitt betitelt „3. Ursachen und Risikofaktoren“Die Ätiologie der IgG4-ROD ist unbekannt, aber es wird angenommen, dass Anomalien der humoralen und zellulären Immunität beteiligt sind.
- B-Zell-Anomalien: Die Wirksamkeit von Rituximab (Anti-CD20) deutet stark auf eine Beteiligung der B-Zellen hin.
- Th2-Zytokine: Eine erhöhte Produktion von IL-4, IL-5 und IL-13 wurde berichtet, begleitet von Eosinophilie und erhöhtem IgE.
- Antigenstimulation: Somatische Hypermutation in der Tränendrüse deutet auf eine lokale Immunantwort hin.
Zu den berichteten Risikofaktoren gehören älteres männliches Geschlecht (systemische Form), atopische Diathese, Asthma und allergische Rhinitis. Patienten mit IgG4-ROD haben möglicherweise ein erhöhtes Risiko für Non-Hodgkin-Lymphome; in den überarbeiteten Kriterien von 2023 warnt Attention II auch vor follikulärem Lymphom und diffusem großzelligen B-Zell-Lymphom (DLBCL) außer MALT 1).
4. Diagnose und Untersuchungsmethoden
Abschnitt betitelt „4. Diagnose und Untersuchungsmethoden“Überarbeitete Diagnosekriterien für IgG4-ROD 2023 1)
Abschnitt betitelt „Überarbeitete Diagnosekriterien für IgG4-ROD 2023 1)“Die Beurteilung erfolgt anhand der folgenden drei Punkte.
| Punkt | Inhalt |
|---|---|
| ① Bildgebung | Vergrößerung der Tränendrüse, Vergrößerung des Trigeminusnervs, Vergrößerung der äußeren Augenmuskeln sowie tumoröse, vergrößerte oder verdickte Läsionen in verschiedenen Augengeweben |
| ② Histopathologie | Ausgeprägtes lymphoplasmazytäres Infiltrat. IgG4+/IgG+ Zellverhältnis ≥ 40 % oder IgG4+ Zellzahl ≥ 50/HPF (×400). Häufig sind Keimzentren zu sehen. |
| ③ Serum-IgG4 | > 135 mg/dL |
- Gesichert: Alle drei Kriterien ①②③ sind erfüllt
- Wahrscheinlich: Zwei Kriterien ①② sind erfüllt
- Verdacht: Zwei Kriterien ①③ sind erfüllt
In der Revision 2023 hinzugefügte Aufmerksamkeit1):
- Aufmerksamkeit I: Achten Sie auf Sehverschlechterung und Gesichtsfeldausfälle durch Optikusneuropathie. Auch eine hypertrophe Pachymeningitis kann eine Optikusneuropathie verursachen.
- Aufmerksamkeit II: Nicht nur MALT-Lymphom, sondern auch follikuläres Lymphom und diffus großzelliges B-Zell-Lymphom (DLBCL) können im Hintergrund einer IgG4-ROD auftreten.
Bildgebende Untersuchungen
Abschnitt betitelt „Bildgebende Untersuchungen“- MRT: Isointens in T1-gewichteten Aufnahmen, hypointens in T2-gewichteten Aufnahmen, homogene Kontrastmittelaufnahme mit Gadolinium.
- FDG-PET/CT: Nützlich zur Erkennung von entfernten und asymptomatischen Läsionen.
Hinweise zu klinischen Untersuchungen
Abschnitt betitelt „Hinweise zu klinischen Untersuchungen“Der Serum-IgG4-Spiegel ist ein Marker für das Ansprechen auf die Behandlung. Es ist jedoch zu beachten, dass 40% der Patienten mit gesicherter IgG4-RD normale Serum-IgG4-Spiegel aufweisen. Zudem kann ein erhöhter IgG4-Spiegel auch bei Pankreaskarzinom, Lymphom und ANCA-assoziierter Vaskulitis auftreten, sodass die Spezifität begrenzt ist.
Obligatorische Untersuchungen sind die Bestimmung des Serum-IgG4-Spiegels, Bildgebung (Kontrast-MRT, CT), Biopsie und histopathologische Untersuchung (einschließlich Immunhistochemie).
Differenzialdiagnose
Abschnitt betitelt „Differenzialdiagnose“Besonders wichtig ist die Abgrenzung zum MALT-Lymphom. Beim MALT-Lymphom ist die IgG4-Färbung in der Regel negativ, es gibt jedoch auch positive Fälle. Die Untersuchung auf IgH-Gen-Rearrangement an der Biopsieprobe ist für die Differenzierung hilfreich.
| Differenzialdiagnosen | Differenzialdiagnostische Punkte |
|---|---|
| MALT-Lymphom | IgH-Gen-Rearrangement-Test. IgG4-Färbung kann positiv sein, Vorsicht geboten. |
| Follikuläres Lymphom / DLBCL | Warnung in den überarbeiteten Kriterien Attention II 2023 1) |
| Sjögren-Syndrom | Anti-Ro/La-Antikörper. Überwiegende Atrophie der Tränen- und Speicheldrüsen. |
| Sarkoidose | ACE-Wert, Thoraxbildgebung, nicht-verkäsende epitheloidzellige Granulome. |
| Granulomatose mit Polyangiitis | ANCA-Serologie |
| Schilddrüsen-Augenerkrankung | Schilddrüsenfunktionstests (T3, T4, TSH). Keine Hypertrophie des Nervus trigeminus. |
| Idiopathische Orbitaentzündung (IOI) | Reklassifizierung durch IgG4-Färbung. 17–60 % der IOI sind IgG4-ROD. |
| Bakterielle oder Pilzinfektion | Akuter Beginn, Schmerz, Fieber. |
5. Standardbehandlung
Abschnitt betitelt „5. Standardbehandlung“Vor der Behandlung ist eine histologische Diagnose zum Ausschluss eines bösartigen Tumors zwingend erforderlich. Bei ausschließlichen Augenveränderungen oder ohne Organbeteiligung außerhalb der Speicheldrüsen ist eine Behandlung nicht immer erforderlich. Allerdings ist eine Behandlung neben kosmetischen Verbesserungen auch bei Sehstörungen wie Sehverschlechterung oder Gesichtsfeldeinschränkung absolut indiziert.
Orale Steroid-Ausschleich-Therapie (erste Wahl)
Abschnitt betitelt „Orale Steroid-Ausschleich-Therapie (erste Wahl)“Beginnen Sie mit 30 mg/Tag Prednisolon (0,6 mg/kg/Tag) und reduzieren Sie die Dosis alle zwei Wochen um 10 %. Setzen Sie die Erhaltungsdosis von 10 mg/Tag für mindestens 3 Monate fort.
Das initiale Ansprechen ist mit 89–100 % sehr gut, aber die Rezidivrate während und nach der Behandlung erreicht bis zu 70 %. Es wurde berichtet, dass die Fortführung einer Erhaltungsdosis von 5 mg/Tag die 3-Jahres-Rezidivrate von 92 % auf 23 % senkt.
Steroid-Pulstherapie (bei Optikusneuropathie)
Abschnitt betitelt „Steroid-Pulstherapie (bei Optikusneuropathie)“Indiziert bei schwerer Sehverschlechterung oder Gesichtsfeldausfällen aufgrund einer Optikusneuropathie. Verabreichen Sie 500 mg Solu-Cortef als intravenöse Infusion einmal täglich über 3 Tage als einen Zyklus und wiederholen Sie 1–3 Zyklen. Eine Übersicht über 44 Fälle von Optikusneuropathie zeigte, dass sich das Sehvermögen in den meisten Fällen mit Steroiden, Rituximab oder dekompressiver Chirurgie besserte, aber die Erholung war bei schweren Fällen mit Lichtwahrnehmung oder weniger schlecht 1).
Rituximab (in Japan nicht von der Krankenkasse erstattet)
Abschnitt betitelt „Rituximab (in Japan nicht von der Krankenkasse erstattet)“Rituximab (Anti-CD20-Antikörper) ist mit einer Ansprechrate von 93 % und einer Rezidivrate von 9 % das wirksamste krankheitsmodifizierende Medikament. Empfohlen werden zwei Infusionen von 1 g im Abstand von 14 Tagen. In Japan ist es nicht von der Krankenkasse erstattet; die Anwendung wird unter Bezugnahme auf Erfahrungen im Ausland erwogen.
Andere Immunsuppressiva
Abschnitt betitelt „Andere Immunsuppressiva“Bei steroidresistenten oder rezidivierenden Fällen können Immunsuppressiva wie Methotrexat, Azathioprin oder Mycophenolatmofetil eingesetzt werden, aber die Evidenz ist begrenzt.
Wenn die Läsion auf die Tränendrüse beschränkt ist oder eine systemische Verabreichung nicht wünschenswert ist, sind auch die Entfernung der Tränendrüse oder die lokale Steroidgabe Optionen.
Rückfälle treten häufig während der Dosisreduktion (Reduzierung von Prednisolon auf weniger als 10 mg/Tag) oder nach Absetzen der Steroide auf. Die Behandlung von Rückfällen umfasst die erneute Gabe oraler Steroide (6–10 Wochen) oder die Hinzunahme krankheitsmodifizierender Medikamente wie Rituximab. Rituximab hat mit 9 % die niedrigste Rückfallrate. Die langfristige Fortführung einer Erhaltungsdosis von 5 mg/Tag kann die 3-Jahres-Rückfallrate von 92 % auf 23 % senken, daher ist ein langfristiger Behandlungsplan wichtig.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Abschnitt betitelt „6. Pathophysiologie und detaillierter Krankheitsmechanismus“Die drei wichtigsten pathologischen Merkmale der IgG4-RD sind:
- Dichte lymphoplasmazelluläre Infiltration
- Storiforme Fibrose
- Obliterierende Phlebitis
Wenn zwei dieser Merkmale (am häufigsten die Kombination von 1 und 2) vorliegen, wird eine IgG4-RD diagnostiziert. Bei der IgG4-ROD werden T-Lymphozyten gefunden, und in Tränendrüsenläsionen kann eine storiforme Fibrose fehlen.
In der Immunhistochemie gelten ein Verhältnis von IgG4-positiven Plasmazellen zu IgG-positiven Zellen ≥ 40 % oder ≥ 50 IgG4-positive Zellen/HPF (×400) als Kriterium. Bei Tränendrüsenerkrankungen wird ein strengeres Kriterium von ≥ 100 IgG4-positiven Zellen/HPF angewendet 1).
Im Zentrum der Pathologie steht eine B-Zell-Anomalie, und die Wirksamkeit von Rituximab bestätigt deren Beteiligung. Die Überproduktion von Th2-Zytokinen (IL-4, IL-5, IL-13) führt zu erhöhten IgG4- und IgE-Spiegeln sowie Eosinophilie. Hinweise auf somatische Hypermutation in der Tränendrüse deuten auf eine lokale Antigenantwort hin.
Bei der IgG4-assoziierten Mikulicz-Krankheit ist eine symmetrische Schwellung der Tränen- und Speicheldrüsen charakteristisch. Aufgrund der begrenzten Zerstörung des Tränendrüsengewebes sind trockene Augensymptome im Gegensatz zum Sjögren-Syndrom oft nicht ausgeprägt. Die Ansprechrate auf eine Steroidtherapie ist im Allgemeinen gut, aber Rückfälle während der Dosisreduktion sind problematisch. In Fällen, die durch eine schwere Optikusneuropathie kompliziert werden, ist eine Erblindung möglich, und die Sehprognose kann schlechter sein als bei orbitalen Low-Grade-Lymphomen.
7. Aktuelle Forschung und zukünftige Perspektiven
Abschnitt betitelt „7. Aktuelle Forschung und zukünftige Perspektiven“Entwicklung der überarbeiteten Diagnosekriterien von 2023 und deren Hintergrund 1)
Abschnitt betitelt „Entwicklung der überarbeiteten Diagnosekriterien von 2023 und deren Hintergrund 1)“Takahira et al. (2024) 1) veröffentlichten eine überarbeitete Version der Diagnosekriterien von 2014, basierend auf einer multizentrischen Studie der Forschungsgruppe für refraktäre Erkrankungen des Ministeriums für Gesundheit, Arbeit und Soziales, mit Ergänzung von Attention I (Warnung vor Optikusneuropathie) und Attention II (Warnung vor Lymphomen außer MALT). Grundlage waren eine multizentrische Studie mit 378 Fällen (Tränendrüse 86 %, äußere Augenmuskeln 21 %, Trigeminusnerv 20 %) und eine Übersicht über 44 Fälle von Optikusneuropathie (Alter 17–86 Jahre, Median 61 Jahre, Geschlechterverhältnis Männer:Frauen 30:14, medianes Serum-IgG4 355 mg/dL). In der Übersicht über 44 Fälle von Optikusneuropathie erholten sich die meisten Fälle mit Steroiden, Rituximab oder dekompressiver Chirurgie, aber schwere Fälle mit Lichtwahrnehmung oder schlechter erholten sich nur unzureichend.
IgG4-ROD nach SARS-CoV-2-Impfung2)
Abschnitt betitelt „IgG4-ROD nach SARS-CoV-2-Impfung2)“Zhang et al. (2024) berichteten über einen Fall von IgG4-ROD nach SARS-CoV-2-Impfung und fassten in einer Literaturübersicht einige Fälle nach Infektion und Impfung zusammen. Davon traten 5 Fälle nach Impfung und 4 Fälle nach Infektion auf. Ein Kausalzusammenhang ist jedoch nicht etabliert, und die Beteiligung einer Immunregulationsstörung ist hypothetisch2).
Assoziation mit SAPHO-Syndrom3)
Abschnitt betitelt „Assoziation mit SAPHO-Syndrom3)“Liu et al. (2025) berichteten über einen Einzelfall einer Assoziation von IgG4-ROD mit SAPHO-Syndrom. Die Beteiligung des TNF-α-Signalwegs ist hypothetisch und nicht als therapeutisches Ziel für beide Erkrankungen etabliert. Die Assoziation beider Erkrankungen ist selten, kann aber einen refraktären Verlauf nehmen3).
Orbitadekompression über transkraniellen Zugang4)
Abschnitt betitelt „Orbitadekompression über transkraniellen Zugang4)“Noda et al. (2021) führten bei einem 63-jährigen Mann mit einem Serum-IgG4-Spiegel von 1.255 mg/dL eine transkranielle Biopsie über den Pterygoidzugang und eine Dekompression der lateralen Orbitawand durch. Am 3. postoperativen Tag verbesserte sich die Sehschärfe von 0,7 LogMAR auf -0,1 LogMAR und der Augeninnendruck normalisierte sich von 31 mmHg auf 15 mmHg. Dies ist als chirurgische Option bei Patienten mit hohem Risiko für eine Steroidtherapie oder bei denen eine schnelle Verbesserung der Sehfunktion erforderlich ist, von Interesse. 4)
8. Literatur
Abschnitt betitelt „8. Literatur“- Takahira M, Goto H, Azumi A. The 2023 revised diagnostic criteria for IgG4-related ophthalmic disease. Jpn J Ophthalmol. 2024;68:293-301.
- Zhang P, Wu Q, Xu X, Chen M. A case of IgG4-related ophthalmic disease after SARS-CoV-2 vaccination: case report and literature review. Frontiers in immunology. 2024;15:1303589. doi:10.3389/fimmu.2024.1303589. PMID:38455056; PMCID:PMC10917890.
- Liu C, Chen T, Wang Y, Wang Q, Hu H, Chen H. SAPHO syndrome complicated by IgG4-related ophthalmic disease: a case report and literature review. Frontiers in immunology. 2025;16:1563542. doi:10.3389/fimmu.2025.1563542. PMID:40342407; PMCID:PMC12058760.
- Noda R, Inoue T, Tsunoda S, Akabane A. Surgical management for IgG4-related ophthalmic disease by a transcranial biopsy combined with extraorbital decompression: illustrative case. Journal of neurosurgery. Case lessons. 2021;1(8):CASE20170. doi:10.3171/CASE20170. PMID:35855308; PMCID:PMC9241348.