母斑症(神経皮膚症候群)は皮膚・神経系・眼に過誤腫性病変を生じる先天性疾患群の総称である。
代表疾患はNF1 ・NF2・結節性硬化症・スタージ・ウェーバー症候群・フォン・ヒッペル・リンドウ病・毛細血管拡張性運動失調症の6疾患である。
NF1 ではLisch結節(虹彩 過誤腫)が成人の90%以上に認められ、診断上重要な眼所見である。SWSでは30〜70%に緑内障 が発生し、先天緑内障 では手術が必須となる。
VHLでは網膜毛細血管腫 が患者の約60%に生じ、1歳から年1回の散瞳 眼底検査 が推奨される。
各疾患の眼合併症は無症状のうちに進行することが多く、定期的な眼科的管理が不可欠である。
分子標的薬(MEK阻害薬、mTOR阻害薬など)が一部疾患で標準治療または新規治療として導入されている。
母斑症(phakomatoses)は、皮膚・中枢神経系・眼に過誤腫性病変を特徴とする先天性疾患群の総称である。神経皮膚症候群(neurocutaneous syndromes)とも呼ばれる。
命名はオランダの眼科医Van der Hoeve(ヴァン・デル・フーフェ)によるもので、ギリシャ語の「phakos(レンズ・斑点)」に由来する。当初は神経線維腫症 ・結節性硬化症・フォン・ヒッペル・リンドウ病の3疾患が含まれ、その後スタージ・ウェーバー症候群・毛細血管拡張性運動失調症が追加された。現在は60以上の症候群が記載されている。
共通する病態基盤は、神経堤細胞の形成・移動・分化の異常である。神経堤細胞は外胚葉由来でシュワン細胞・メラノサイトなど多様な細胞を産生するため、神経・皮膚・眼の多臓器に病変が生じる。関与するシグナル経路としてRAS・MAPK/MEK・mTOR・PI3K/AKT・GNAQ・VHL-HIF経路が知られている。
主要6疾患の発生頻度は以下の通りである。
疾患 発生頻度(人に1人) NF1 (神経線維腫症1型 )3,000〜5,000 結節性硬化症(TSC ) 6,000〜10,000 SWS(スタージ・ウェーバー症候群) 20,000〜50,000 VHL(フォン・ヒッペル・リンドウ病) 36,000 NF2(神経線維腫症 2型) 25,000〜100,000 AT(毛細血管拡張性運動失調症) 88,000〜100,000未満
Q
母斑症にはどのような疾患が含まれるか?
A
代表的な6疾患はNF1 ・NF2・結節性硬化症・スタージ・ウェーバー症候群・フォン・ヒッペル・リンドウ病・毛細血管拡張性運動失調症である。いずれも遺伝子変異に起因し、神経・皮膚・眼の多臓器に病変を生じる。現在は60以上の症候群が母斑症の範疇に含まれる。
An Update on Multimodal Ophthalmological Imaging of
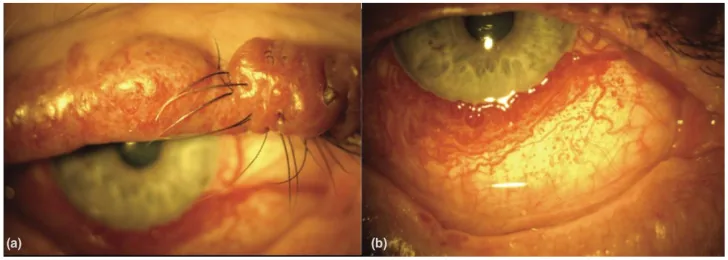
Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Port-wine stain of the upper lid with nodularity in a patient with Sturge–Weber syndrome. (b) Diffuse conjunctival vascularity in a patient with Sturge–Weber syndrome. From [15].
各疾患で眼合併症の種類が異なり、自覚症状も多様である。
NF1 視神経膠腫 )が進行すると、視力 低下・色覚喪失・視野欠損 が生じる。蔓状神経線維腫では眼球突出 をきたすことがある。結節性硬化症 :網膜星細胞過誤腫 は通常無症状である。黄斑 や視神経乳頭 を侵した場合に視力 障害をきたす。SWS :先天緑内障 を伴う場合、角膜 混濁・流涙・羞明 が生じる。後期発症の緑内障 では無痛性の進行性視野欠損 が起こる。VHL :早期は無症状。網膜毛細血管腫 が進行すると滲出性変化・黄斑浮腫 ・輪状白斑が出現し、視力 低下をきたす。AT :視力 は通常維持される。眼球運動障害 (眼振 ・眼球運動失行 )が主体である。
Lisch結節(リッシュ結節) :NF1 で最も頻度の高い眼所見。淡褐色・境界鮮明・ドーム状の小結節(1〜2mm未満)が虹彩 に多発する。年齢別有病率は3歳未満5%・3〜4歳42%・5〜6歳55%・21歳以上100%と年齢とともに増加し、NF1 の診断基準(2個以上)に含まれる。日本人では虹彩 色が茶系のため、細隙灯による精査が重要である。視路腫瘍(視神経膠腫 ) :NF1 患者の約15〜25%に合併。低悪性度毛様細胞性星細胞腫が多く、無症候性のことが多い。進行すると視神経萎縮 ・視力 障害・視野障害をきたす。視交叉 浸潤例もある。蔓状神経線維腫 :NF1 患者の10%未満に発生。眼瞼の「S字型変形」が特徴で、触診で「虫袋(bag of worms)」様の感触がある。眼球突出 ・斜視 ・弱視 ・先天緑内障 の原因となる。蝶形骨翼形成不全 :眼窩 骨壁の先天性欠損で、拍動性眼球突出 を生じることがある。緑内障 NF1 の1〜2%に発生。先天性(片眼性)と遅発症の2タイプがある。その他 :角膜 有髄神経の著明化、脈絡膜 肥厚、網膜 有髄神経。
視神経鞘髄膜腫 NF1 の視路膠腫とは対照的な腫瘍型である。白内障 白内障 がみられる。網膜 ・RPE 過誤腫網膜 上膜露出性角膜 炎 :両側聴神経腫瘍による第5・第7脳神経障害が顔面しびれ・複視 ・兎眼 を引き起こし、続発する。Lisch結節はまれである。
網膜星細胞過誤腫 網膜 脱色素病変網膜 血管異常硝子体出血 ・増殖性硝子体網膜症 ・網膜剥離 の原因となりうる。その他 :眼瞼血管線維腫、虹彩 脱色素斑、非定型的脈絡膜 欠損。
三主徴は(1)三叉神経 領域の顔面血管腫、(2)同側頭蓋内血管腫、(3)同側の緑内障 または脈絡膜 血管腫である。
緑内障 先天緑内障 (生後〜4歳)が約60%を占め、牛眼・角膜 混濁・巨大角膜 をきたす。病因は隅角 発育異常・上強膜 静脈圧上昇・脈絡膜 血管腫の関与が考えられている。眼瞼血管腫の存在下で高頻度に発生する。脈絡膜 血管腫眼底検査 では同定が困難である。眼底は「トマトケチャップ状」外観を呈する。通常は増大傾向を示さないが、滲出性変化・滲出性網膜剥離 をきたすことがある。その他 :結膜 ・上強膜 ・虹彩 の血管拡張蛇行。
網膜毛細血管腫 (血管芽腫)網膜 耳側周辺部に好発し、拡張蛇行した輸出入血管を伴う赤橙色結節として観察される。平均発症年齢は25歳で、通常30歳までに出現する。進行すると滲出性変化・網膜 出血→黄斑浮腫 ・輪状白斑→視力 低下をきたす。
さらに進行すると硝子体出血 ・牽引性網膜剥離 ・血管新生緑内障 →失明に至りうる。
蛍光眼底造影 では輸入動脈への色素流入→輸出静脈灌流の所見を示し、腫瘤部で早期より著明な色素漏出がみられる。
球結膜 毛細血管拡張 :最も一般的な眼所見。通常5〜8歳までに発生し、患者の80〜90%にみられる1) 。眼球運動障害 眼振 ・眼球運動失行 (oculomotor apraxia)・サッケード 異常・輻輳と調節の異常・斜視 が認められる。視力 は通常維持される。
Q
Lisch結節は年齢によってどのように変化するか?
A
NF1 に伴うLisch結節は年齢とともに出現頻度が上昇する。3歳未満では5%にとどまるが、21歳以上ではほぼ100%に認められる。若年では細隙灯検査でも検出困難な場合があり、年齢を考慮した解釈が必要である。
各疾患の遺伝形式・原因遺伝子・染色体座位を以下に示す。
疾患 遺伝形式 原因遺伝子 染色体座位 NF1 常染色体優性 NF1 17q11.2 NF2 常染色体優性 NF2 22q11.1-q13.1 TSC 常染色体優性 TSC1/TSC2 9q34/16p13 VHL 常染色体優性 VHL 3p25-26 SWS 孤発性(体細胞モザイク) GNAQ 9q21 AT 常染色体劣性 ATM 11q22
各疾患の遺伝的特徴は以下の通りである。
NF1 2) 。NF2 :マーリン(merlin)タンパク質の機能喪失。シュワン細胞・軟膜細胞に主に発現する腫瘍抑制因子で、欠損するとシュワン腫・髄膜腫・上衣腫が生じる。TSC 1) 。ハマルチン/ツベリンはmTOR経路を直接阻害する2) 。SWS :孤発性でGNAQ遺伝子の体細胞モザイク変異が原因1) 。三叉神経 第1枝(V1)領域全体のポートワイン母斑(PWB)は眼科・神経学的合併症のリスクが高い。VHL :浸透率はほぼ100%で、約20%がde novo変異。pVHLはHIF-αのユビキチン化・プロテアソーム分解に関与する2) 。Type 1(褐色細胞腫なし)が約80%、Type 2(褐色細胞腫あり)が約20%2) 。AT :常染色体劣性遺伝 でATMキナーゼはDNA二重鎖切断修復・ゲノム安定性維持に関与する1) 。悪性腫瘍(リンパ系)リスクおよび放射線感受性が著しく高い。
NF1 の診断基準(NIH基準・日本皮膚科学会2008年)では、以下7項目のうち2つ以上を満たすことで確定診断される。
6個以上のカフェオレ斑(思春期前:最大径5mm以上、思春期後:15mm以上)
2個以上の神経線維腫またはびまん性神経線維腫
腋窩・鼠径部の雀卵斑様色素斑
視神経 グリオーマ 2個以上のLisch虹彩 結節
特徴的骨病変(蝶形骨異形成など)
第一度近親者に同症
眼科的検査の役割は以下の通りである。
細隙灯顕微鏡 :Lisch結節の検出(若年では困難なことがある)。前眼部OCT ・超音波生体顕微鏡 MRI :視路腫瘍の最良の診断法。視交叉 ・視索の評価に必須。視野検査 光干渉断層計 (OCT )網膜神経線維層 菲薄化の評価。VEP (視覚誘発電位 )視神経 障害の早期検出。定期検査間隔 :Lisch結節のみ→年1回。視神経膠腫 を有する場合→3か月に1回。
臨床基準では両側前庭神経シュワン腫が特徴的所見である。皮膚症状が乏しいためNF1 より診断困難な場合がある。遺伝子検査では90%以上の変異が検出される。CT・MRIで両側聴神経腫瘍を確認する。
主要所見2つ、または主要所見1つ+副所見2つ以上で確定診断される。TSC1/TSC2病原性変異の同定は独立した診断基準として認められている1) 。
網膜星細胞過誤腫 の評価には眼底検査 ・蛍光眼底造影 ・OCT を用いる。鑑別診断で最も重要なのは網膜芽細胞腫 である。小児期に石灰化を伴わない・栄養血管が乏しい・全身症状を伴うという特徴から鑑別が可能だが、桑の実状腫瘍との鑑別は困難なことがある。
Roach診断スケールでは、顔面ポートワイン母斑・眼圧 上昇・軟膜血管腫のうち少なくとも2つを満たすことが基準とされる1) 。脈絡膜 血管腫の評価にはED-OCT ・MRIが有用である。頭部CTでは脳皮質内の石灰化が認められる。
緑内障 のモニタリングには眼圧測定 ・視神経 評価・視野検査 を定期的に行う。乳幼児では細隙灯検査に加え、麻酔下での眼底検査 ・眼圧測定 が必要となることがある。
遺伝子検査ではほぼ100%でVHL変異を検出でき、現在の確定診断の主流である。散瞳 眼底検査 は1歳から年1回の実施が推奨される。周辺部の赤橙色球状腫瘍+拡張蛇行血管が特徴的所見である。蛍光眼底造影 は小さな周辺部血管芽腫の検出に有用で、OCT は小さな病変の評価に用いる。
臨床診断では運動失調・球結膜 毛細血管拡張・眼球運動異常の三徴が重要である。血清AFP上昇・CA125上昇・ATMタンパク質欠損(ウェスタンブロット)が検査所見として認められる。球結膜 毛細血管拡張は本疾患に特異的な所見(pathognomonic)である。MRIでは後頭蓋窩のびまん性小脳萎縮(特に虫部・半球)を示す1) 。
Lisch結節 :無症状の過誤腫であり治療は不要。視神経膠腫 視交叉 浸潤例では化学療法が適応となる。蔓状神経線維腫 :外科的全摘出は困難で再発しやすい。進行例では眼窩内容除去術 が必要になることがある。切除不能例にはセルメチニブ(MEK阻害薬)が新規治療として使用される1) 。緑内障 緑内障 治療に準じた管理を行う。
前庭神経鞘腫に対しては外科的切除が基本で、3cm未満では65%で聴力温存が可能とされる1) 。露出性角膜 炎は人工涙液・角膜 保護眼鏡・必要に応じて瞼板 縫合で対処する。
網膜星細胞過誤腫 網膜 血管異常(動脈瘤様拡張・動静脈奇形)硝子体出血 ・網膜剥離 のリスクがあるため予防的光凝固を行う。硝子体出血 ・網膜剥離 が発生した場合は硝子体手術 を検討する。てんかん :乳児の部分発作・点頭てんかんにはビガバトリンが第一選択とされる1) 。SEGA(上衣下巨細胞性星細胞腫)は神経外科的切除の適応となる1) 。
緑内障 の治療
先天緑内障 (早期発症型)線維柱帯切開術 ・隅角 切開術が選択される。脈絡膜 剥離・出血に対する十分な注意が必要であり、上強膜 静脈圧上昇から通常より合併症リスクが高い。薬物治療への抵抗性が高い傾向がある。後期発症型 :まず薬物治療を行い、無効な場合は手術を検討する。
脈絡膜 血管腫の治療
滲出性変化が生じた場合→光凝固
光凝固無効または滲出性網膜剥離 →放射線治療(総線量20グレイ程度)。胞状網膜剥離 の復位・血管腫縮小が期待できる。
難治例→硝子体手術
光線力学療法(PDT )・外照射放射線療法も選択肢となりうる3) 。
早期治療が原則である。
光凝固(第一選択) :アルゴンレーザーまたはダイレーザーを用い、血管腫周囲の網膜 と栄養血管を密に凝固後、本体を直接凝固する。2乳頭径以上の腫瘍では流入動脈・周囲網膜 を凝固後に腫瘍を直接凝固する。冷凍凝固 強膜 的に施行する。硝子体手術 網膜剥離 ・増殖性変化が生じた場合に検討する。末期例 :血管新生緑内障 に対する管理が必要となる。家族歴がある場合は家族全員の眼底検査 を実施し、早期治療で予後良好が期待できる。
結膜 毛細血管拡張に特異的な治療はない。免疫不全に対しては予防的抗菌薬・免疫グロブリン静脈内投与(IVIg )が行われる1) 。運動失調は対症療法が中心である。
Q
VHLの網膜血管腫はいつから検査を受けるべきか?
A
VHLでは1歳から年1回の散瞳 眼底検査 が推奨されている。VHL遺伝子変異が確認されている患者の家族も全員が眼底検査 を受けることが重要で、早期治療により良好な視機能予後が期待できる。
各疾患の分子レベルの発症機序を以下に示す。
NF1・TSC(mTOR経路)
NF1 (ニューロフィブロミン)2) 。
TSC (ハマルチン/ツベリン)2) 。TSC2変異はTSC1変異よりも重症の表現型をとる1) 。
VHL(HIF経路)
pVHL :E3ユビキチンリガーゼ複合体(elongin B/C・Cullin 2・RBX1)の構成要素。正常酸素下ではプロリル水酸化酵素がHIF-αを水酸化→pVHLが認識→ユビキチン化→プロテアソーム分解。
VHL変異 :偽低酸素状態が恒常的に生じる→HIF-α蓄積→HIF-1βとのヘテロダイマー形成→VEGF・赤血球産生・代謝・細胞増殖関連遺伝子の転写活性化→血管腫瘍形成2) 。
NF2(マーリン) :腫瘍抑制タンパク質でシュワン細胞・軟膜細胞に発現。変異によりシュワン腫・髄膜腫・上衣腫が生じる。SWS(GNAQ体細胞モザイク変異) :Gタンパク質関連膜貫通シグナル伝達の異常。胚における変異時期・場所によって三叉神経 領域・頭蓋内・眼における発現パターンが異なる1) 。GNAQはSWS・KTS・PPV に共通する分子基盤でもある3) 。AT(ATMキナーゼ) :DNA二重鎖切断修復・ゲノム安定性維持に関与する腫瘍抑制因子。変異→DNA修復障害→癌素因・放射線感受性の著しい増大・免疫不全1) 。
Chevalierら(2021)は母斑症と内分泌腫瘍の関連を検討し、NF1 ・TSC ・VHLのシグナル経路(RAS-PI3K-Akt-mTOR経路、HIF経路)が多発性内分泌腫瘍の発生に共通して関与することを報告した2) 。
Q
なぜ母斑症は多臓器に病変を生じるのか?
A
母斑症の共通病態基盤は、神経堤細胞の形成・移動・分化の異常である。神経堤細胞は外胚葉由来でシュワン細胞・メラノサイトなど多様な細胞を産生するため、神経・皮膚・眼の多臓器に病変が及ぶ。加えてRAS-mTOR経路やVHL-HIF経路など共通シグナル経路の変異が、臓器を超えた腫瘍発生を促進する。
切除不能な蔓状神経線維腫に対してFDA承認を受けた分子標的薬である。NF1 のMAPK経路の過活性を標的とする1) 。
NF2関連シュワン腫・進行性腫瘍に対してINTUITT-NF2コンソーシアムでの臨床試験が実施されており、有望な結果が報告されている1) 。
VHL関連淡明細胞腎細胞癌・膵神経内分泌腫瘍を対象としたフェーズ2試験が進行中である。
Chevalierら(2021)によるMK6482の試験では、膵神経内分泌腫瘍の64%に客観的奏効が得られ、12か月無増悪生存率は98.3%と報告された2) 。
TSC 関連腎血管筋脂肪腫・SEGAに対するEXIST試験で形態学的奏効が確認されており、内分泌腫瘍への応用も期待されている2) 。
GNAQ遺伝子変異がSWS・クリッペル・トレノネー症候群(KTS)・色素血管性母斑症(PPV )に共通の分子基盤であることが判明した3) 。これらの重複症例では脈絡膜メラノーマ のリスクがあるため、年1〜2回の散瞳 眼底検査 が推奨されている3) 。
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
記事の全文をコピーして、お好みのAIに貼り付けて質問できます
下のAIを開いて、チャット欄に貼り付け(ペースト)してください