斑痣 性错构瘤病(神经皮肤综合征)是一组先天性疾病的统称,其特征是在皮肤、神经系统和眼睛中发生错构瘤性病变。
代表性疾病包括NF1 、NF2、结节性硬化症、斯特奇-韦伯综合征、冯·希佩尔-林道病和毛细血管扩张性共济失调症这六种疾病。
在NF1 中,超过90%的成人可见Lisch结节(虹膜 错构瘤),这是诊断中重要的眼部表现。
在SWS中,30%至70%的患者发生青光眼 ,先天性青光眼 需要手术治疗。
在VHL中,约60%的患者出现视网膜毛细血管瘤 ,建议从1岁起每年进行一次散瞳 眼底检查 。
每种疾病的眼部并发症常在无症状时进展,因此定期的眼科管理至关重要。
分子靶向药物(如MEK抑制剂、mTOR抑制剂)已被引入作为部分疾病的标准治疗或新疗法。
斑痣 性错构瘤病是一组以皮肤、中枢神经系统和眼睛的错构瘤性病变为特征的先天性疾病的总称。也称为神经皮肤综合征。
该命名由荷兰眼科医生Van der Hoeve提出,源自希腊语“phakos”(透镜或斑点)。最初包括神经纤维瘤病 、结节性硬化症和冯·希佩尔-林道病,随后加入了斯特奇-韦伯综合征和毛细血管扩张性共济失调。目前已有超过60种综合征被描述。
共同的病理基础是神经嵴细胞的形成、迁移和分化异常。由于神经嵴细胞来源于外胚层,产生施万细胞、黑素细胞等多种细胞,因此神经、皮肤和眼睛等多个器官会出现病变。已知涉及的信号通路包括RAS、MAPK/MEK、mTOR、PI3K/AKT、GNAQ和VHL-HIF通路。
六大主要疾病的发病率如下。
疾病 发病率(每X人中1例) NF1 (神经纤维瘤病1型 )3,000~5,000 结节性硬化症(TSC ) 6,000~10,000 SWS(斯特奇-韦伯综合征) 20,000~50,000 VHL(冯·希佩尔-林道病) 36,000 NF2(神经纤维瘤病 2型) 25,000~100,000 毛细血管扩张性共济失调症(AT) 88,000至100,000以下
Q
母斑症包括哪些疾病?
A
代表性的6种疾病是NF1 、NF2、结节性硬化症、斯特奇-韦伯综合征、冯·希佩尔-林道病和毛细血管扩张性共济失调症。所有这些疾病均由基因突变引起,并在神经、皮肤和眼等多器官产生病变。目前有60多种综合征被归入母斑症的范畴。
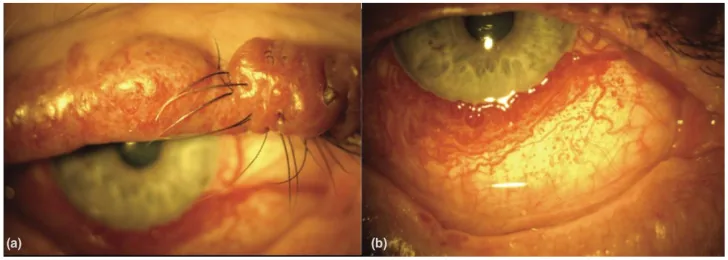
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) 斯特奇-韦伯综合征患者上眼睑葡萄酒色斑伴结节。 (b) 斯特奇-韦伯综合征患者弥漫性结膜 血管。摘自[15]。
各疾病的眼部并发症类型不同,自觉症状也多种多样。
NF1 视神经胶质瘤 )进展时,会出现视力 下降、色觉丧失和视野缺损 。丛状神经纤维瘤可能导致眼球突出 。结节性硬化症 :视网膜星形细胞错构瘤 通常无症状。当累及黄斑 或视盘时,会导致视力 障碍。SWS :伴有先天性青光眼 时,会出现角膜 混浊、流泪和畏光 。迟发性青光眼 导致无痛性进行性视野缺损 。VHL :早期无症状。视网膜毛细血管瘤 进展时出现渗出性改变、黄斑水肿 、环状白斑,导致视力 下降。AT :视力 通常保持正常。主要表现为眼球运动障碍 (眼球震颤 、眼球运动失用 )。
Lisch结节 :NF1 最常见的眼部表现。虹膜 上多发淡褐色、边界清晰、圆顶状的小结节(<1–2 mm)。年龄别患病率:<3岁5%,3–4岁42%,5–6岁55%,≥21岁100%,随年龄增长而增加,并纳入NF1 诊断标准(≥2个)。日本人虹膜 颜色偏棕,因此裂隙灯 下仔细检查很重要。视路肿瘤(视神经胶质瘤 ) :约15–25%的NF1 患者合并。多为低级别毛细胞星形细胞瘤,常无症状。进展时可导致视神经萎缩 、视力 障碍和视野缺损 。可能侵犯视交叉 。丛状神经纤维瘤 :发生于不到10%的NF1 患者。特征为眼睑的“S形变形”,触诊时有“虫袋”样感觉。可导致眼球突出 、斜视 、弱视 和先天性青光眼 。蝶骨翼发育不全 :眼眶 骨壁的先天性缺损 ,可能引起搏动性眼球突出 。青光眼 NF1 患者。有先天性(单眼)和迟发性两种类型。其他 :角膜 有髓神经纤维明显、脉络膜 增厚、视网膜有髓神经纤维 。
视神经鞘脑膜瘤 NF1 的视路胶质瘤 形成对比的肿瘤类型。白内障 白内障 。视网膜 /RPE 错构瘤视网膜前膜 暴露性角膜 炎 :双侧听神经瘤导致第V、第VII脑神经功能障碍,引起面部麻木、复视 、兔眼 ,继发暴露性角膜 炎。Lisch结节罕见。
视网膜星形细胞错构瘤 视网膜 脱色素病变视网膜 血管异常玻璃体出血 、增生性玻璃体视网膜病变 和视网膜脱离 。其他 :眼睑血管纤维瘤、虹膜 脱色素斑、非典型脉络膜 缺损 。
三大主要特征为:(1)三叉神经 分布区的面部血管瘤,(2)同侧颅内血管瘤,(3)同侧青光眼 或脉络膜 血管瘤。
青光眼 先天性青光眼 (出生至4岁)约占60%,导致牛眼、角膜 混浊和大角膜 。病因被认为涉及房角 发育异常、巩膜 上静脉压升高和脉络膜 血管瘤。在眼睑血管瘤存在时发生频率高。脉络膜 血管瘤眼底检查 难以识别。眼底呈“番茄酱样”外观。通常无增大趋势,但可引起渗出性改变和渗出性视网膜脱离 。其他 :结膜 、上巩膜 和虹膜 的血管扩张迂曲。
视网膜毛细血管瘤 (血管母细胞瘤)视网膜 颞侧周边部,表现为伴有扩张迂曲的输入和输出血管的橙红色结节。平均发病年龄为25岁,通常在30岁前出现。进展可引起渗出性改变、视网膜 出血、黄斑水肿 、环状白斑和视力 下降。
进一步进展可导致玻璃体出血 、牵拉性视网膜脱离 、新生血管性青光眼 和失明。
荧光眼底造影显示染料流入输入动脉→输出静脉灌注,肿瘤区域早期出现明显染料渗漏。
球结膜 毛细血管扩张 :最常见的眼部表现。通常在5~8岁前出现,见于80%~90%的患者1) 。眼球运动障碍 眼球震颤 、眼球运动失用 、扫视 异常、辐辏和调节异常、斜视 。视力 通常保持正常。
Q
Lisch结节随年龄如何变化?
A
与NF1 相关的Lisch结节随年龄增长出现频率增加。3岁以下仅5%出现,但21岁以上几乎100%可见。在年轻患者中,即使裂隙灯 检查也可能难以检测,因此需要考虑年龄因素进行解读。
各疾病的遗传方式、致病基因和染色体位点如下所示。
疾病 遗传方式 致病基因 染色体位点 NF1 常染色体显性 NF1 17q11.2 NF2 常染色体显性 NF2 22q11.1-q13.1 TSC 常染色体显性 TSC1/TSC2 9q34/16p13 VHL 常染色体显性 VHL 3p25-26 SWS 散发性(体细胞嵌合) GNAQ 9q21 AT 常染色体隐性 ATM 11q22
各疾病的遗传特征如下。
NF1 2) 。NF2 :梅林蛋白功能丧失。主要表达于施万细胞和脑膜细胞的肿瘤抑制因子,其缺失导致施万瘤、脑膜瘤和室管膜瘤。TSC 1) 。错构蛋白/结节蛋白直接抑制mTOR通路2) 。SWS :散发性,由GNAQ基因体细胞嵌合突变引起1) 。累及三叉神经 第一支(V1)整个区域的葡萄酒色斑(PWB)具有较高的眼部和神经系统并发症风险。VHL :外显率几乎100%,约20%为新发突变。pVHL参与HIF-α的泛素化和蛋白酶体降解2) 。1型(无嗜铬细胞瘤)约占80%,2型(有嗜铬细胞瘤)约占20%2) 。AT :常染色体隐性遗传 ,ATM激酶参与DNA双链断裂修复和基因组稳定性维持1) 。恶性肿瘤(淋巴系统)风险和放射敏感性显著增高。
NF1 的诊断标准(NIH标准、日本皮肤科学会2008年)要求满足以下7项中的2项或以上方可确诊。
6个或更多咖啡牛奶斑(青春期前:最大直径≥5mm;青春期后:≥15mm)
2个或更多神经纤维瘤或1个弥漫性神经纤维瘤
腋窝或腹股沟区雀斑样色素沉着
视神经胶质瘤 2个或更多Lisch虹膜 结节
特征性骨病变(如蝶骨发育不良)
一级亲属患有相同疾病
眼科检查的作用如下。
裂隙灯 显微镜眼前节OCT 和超声生物显微镜 :有助于辅助检测Lisch结节。MRI :视路肿瘤的最佳诊断方法。对视交叉 和视束的评估至关重要。视野检查 光学相干断层扫描 (OCT )视网膜神经纤维层 变薄。VEP (视觉诱发电位 )视神经 损伤。定期检查间隔 :仅有Lisch结节→每年1次。伴有视神经胶质瘤 →每3个月1次。
临床标准中,双侧前庭神经鞘瘤是特征性表现。由于皮肤症状不明显,诊断可能比NF1 更困难。基因检测可检出90%以上的突变。CT/MRI可确认双侧听神经瘤。
确诊需要满足2项主要特征,或1项主要特征加2项及以上次要特征。TSC1/TSC2致病性变异的鉴定被认可为独立的诊断标准1) 。
视网膜星形细胞错构瘤 的评估使用眼底检查 、荧光眼底造影和OCT 。最重要的鉴别诊断是视网膜母细胞瘤 。根据儿童期无钙化、营养血管稀少、伴有全身症状的特点可以进行鉴别,但与桑葚状肿瘤的鉴别有时较为困难。
Roach诊断量表要求至少满足面部葡萄酒色斑、眼压 升高、软脑膜血管瘤中的两项1) 。增强深度OCT 和MRI对脉络膜 血管瘤的评估有用。头部CT显示脑皮质内钙化。
青光眼 的监测需要定期进行眼压测量 、视神经 评估和视野检查 。婴幼儿可能需要在麻醉下进行裂隙灯 检查、眼底检查 和眼压测量 。
基因检测几乎100%能检测出VHL突变,是目前确诊的主流方法。建议从1岁起每年进行一次散瞳 眼底检查 。周边部橙红色球形肿瘤伴扩张迂曲血管是特征性表现。荧光眼底造影有助于发现小的周边部血管母细胞瘤,OCT 用于评估小病变。
临床诊断中,共济失调、球结膜 毛细血管扩张和眼球运动异常三联征很重要。实验室检查可见血清AFP升高、CA125升高和ATM蛋白缺失(Western blot)。球结膜 毛细血管扩张是本病的特征性表现(pathognomonic)。MRI显示后颅窝弥漫性小脑萎缩,尤其是蚓部和半球1) 。
Lisch结节 :无症状的错构瘤,无需治疗。视神经胶质瘤 视交叉 浸润病例适用化疗。蔓状神经纤维瘤 :手术完全切除困难且易复发。进展病例可能需要眼眶 内容物剜除术。不可切除病例可使用司美替尼(MEK抑制剂)作为新疗法1) 。青光眼 青光眼 治疗进行管理。
前庭神经鞘瘤以手术切除为基本治疗,小于3cm的肿瘤中65%可保留听力1) 。暴露性角膜 炎使用人工泪液、角膜 保护眼镜,必要时行睑板 缝合处理。
视网膜星形细胞错构瘤 视网膜 血管异常(动脉瘤样扩张、动静脉畸形)玻璃体出血 和视网膜脱离 的风险,需进行预防性光凝治疗。若发生玻璃体出血 或视网膜脱离 ,考虑玻璃体 手术。癫痫 :婴儿局灶性发作和婴儿痉挛症的首选治疗为氨己烯酸1) 。SEGA(室管膜下巨细胞星形细胞瘤)需神经外科手术切除1) 。
青光眼 的治疗
先天性青光眼 (早发型)小梁切开术 或房角 切开术。需注意脉络膜 脱离和出血,由于巩膜 上静脉压升高,并发症风险高于常规。对药物治疗往往耐药。晚期发病型 :首先进行药物治疗,无效时考虑手术。
脉络膜 血管瘤的治疗
早期治疗是原则。
光凝(首选) :使用氩激光或染料激光,先致密凝固血管瘤周围的视网膜 和滋养血管,然后直接凝固瘤体。对于大于2个视盘直径的肿瘤,先凝固流入动脉和周围视网膜 ,再直接凝固肿瘤。冷冻凝固 巩膜 施行。玻璃体切除术 视网膜脱离 或出现增殖性改变时考虑。终末期病例 :需要管理新生血管性青光眼 。如有家族史,应对所有家庭成员进行眼底检查 ,早期治疗可望获得良好预后。
结膜 毛细血管扩张没有特异性治疗。对于免疫缺陷,进行预防性抗生素和静脉注射免疫球蛋白(IVIg )治疗1) 。共济失调以对症治疗为主。
Q
VHL视网膜血管瘤应从何时开始检查?
A
对于VHL,建议从1岁开始每年进行一次散瞳 眼底检查 。已确认VHL基因突变的患者的所有家庭成员也应接受眼底检查 ,早期治疗有望获得良好的视功能预后。
各疾病的分子水平发病机制如下所示。
NF1与TSC(mTOR通路)
NF1 (神经纤维蛋白)2) 。
TSC (错构蛋白/结节蛋白)2) 。TSC2突变比TSC1突变导致更严重的表型1) 。
VHL(HIF通路)
pVHL :E3泛素连接酶复合体(elongin B/C、Cullin 2、RBX1)的组成部分。在正常氧条件下,脯氨酰羟化酶将HIF-α羟化→pVHL识别→泛素化→蛋白酶体降解。
VHL突变 :持续出现假性缺氧状态→HIF-α积累→与HIF-1β形成异二聚体→VEGF、红细胞生成、代谢和细胞增殖相关基因的转录激活→血管肿瘤形成2) 。
NF2(merlin) :肿瘤抑制蛋白,在施万细胞和软脑膜细胞中表达。突变导致施万瘤、脑膜瘤和室管膜瘤。SWS(GNAQ体细胞嵌合突变) :G蛋白相关跨膜信号传导异常。胚胎中突变的时间和位置导致三叉神经 区域、颅内和眼部的表达模式不同1) 。GNAQ也是SWS、KTS和PPV 的共同分子基础3) 。AT(ATM激酶) :参与DNA双链断裂修复和基因组稳定性维持的肿瘤抑制因子。突变→DNA修复障碍→癌症易感性、放射敏感性显著增加和免疫缺陷1) 。
Chevalier等人(2021)研究了母斑病与内分泌肿瘤的关联,报告称NF1 、TSC 和VHL的信号通路(RAS-PI3K-Akt-mTOR通路、HIF通路)共同参与多发性内分泌肿瘤 的发生2) 。
Q
为什么母斑病会在多个器官产生病变?
A
母斑病的共同病理基础是神经嵴细胞的形成、迁移和分化异常。神经嵴细胞来源于外胚层,产生施万细胞、黑色素细胞等多种细胞,因此病变累及神经、皮肤、眼等多个器官。此外,RAS-mTOR通路和VHL-HIF通路等共同信号通路的突变促进了跨器官的肿瘤发生。
一种获得FDA批准用于不可切除的丛状神经纤维瘤的分子靶向药物。它靶向NF1 中MAPK通路的过度激活1) 。
针对NF2相关施万瘤和进展性肿瘤的临床试验由INTUITT-NF2联盟进行,已报告有希望的结果1) 。
针对VHL相关透明细胞肾细胞癌和胰腺神经内分泌肿瘤的2期试验正在进行中。
Chevalier等人(2021)的MK6482试验中,64%的胰腺神经内分泌肿瘤获得客观缓解,12个月无进展生存率为98.3% 2) 。
针对TSC 相关肾血管平滑肌脂肪瘤和SEGA的EXIST试验证实了形态学缓解,其在内分泌肿瘤中的应用也值得期待 2) 。
GNAQ基因突变已被确定为SWS、克利佩尔-特雷诺内综合征(KTS)和色素血管性斑痣 性错构瘤病(PPV )的共同分子基础 3) 。这些重叠病例存在脉络膜黑色素瘤 的风险,因此建议每年进行1-2次散瞳 眼底检查 3) 。
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
复制全文后,可以粘贴到你常用的 AI 助手中提问。
打开下面的 AI 助手,并把复制的内容粘贴到聊天框。