Fakomatozlar (phakomatoses), deri, merkezi sinir sistemi ve gözde hamartomatöz lezyonlarla karakterize konjenital hastalık grubudur. Nörokütanöz sendromlar (neurocutaneous syndromes) olarak da adlandırılır.
Adlandırma Hollandalı göz doktoru Van der Hoeve tarafından yapılmış olup, Yunanca «phakos» (mercek/benek) kelimesinden türetilmiştir. Başlangıçta nörofibromatozis, tüberoz skleroz ve von Hippel-Lindau hastalığı olmak üzere üç hastalığı içerirken, daha sonra Sturge-Weber sendromu ve ataksi telenjiektazi eklenmiştir. Günümüzde 60’tan fazla sendrom tanımlanmıştır.
Ortak patolojik temel, nöral krest hücrelerinin oluşumu, göçü ve farklılaşmasındaki anormalliktir. Nöral krest hücreleri ektoderm kaynaklı olup Schwann hücreleri, melanositler gibi çeşitli hücreleri üretir; bu nedenle sinir, deri ve göz gibi çoklu organlarda lezyonlar oluşur. İlgili sinyal yolları RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ ve VHL-HIF yollarını içerir.
Başlıca altı hastalığın görülme sıklığı aşağıdaki gibidir:
Altı ana hastalık NF1, NF2, tüberoz skleroz, Sturge-Weber sendromu, von Hippel-Lindau hastalığı ve ataksi telenjiektazidir. Bunların tümü gen mutasyonlarından kaynaklanır ve sinir, cilt ve göz gibi çoklu organlarda lezyonlara yol açar. Günümüzde 60’tan fazla sendrom fakomatoz kategorisine dahil edilmektedir.
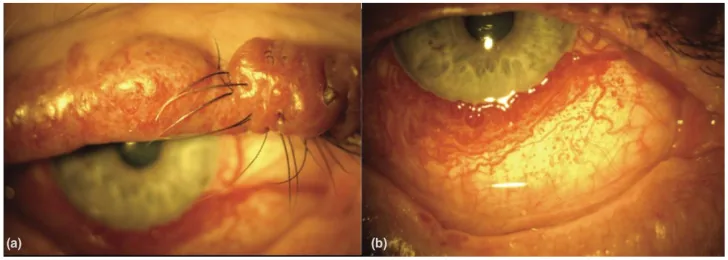
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Sturge–Weber sendromlu bir hastada nodüler üst göz kapağı porto şarabı lekesi. (b) Sturge–Weber sendromlu bir hastada yaygın konjonktival vaskülarite. [15]‘ten.
Her hastalıkta oküler komplikasyonların türü farklıdır ve subjektif semptomlar da çeşitlidir.
NF1: Görme yolu tümörü (optik gliom) ilerledikçe görme azalması, renk görme kaybı ve görme alanı defekti oluşur. Pleksiform nörofibromda proptozis görülebilir.
Tüberoskleroz: Retinal astrositik hamartom genellikle asemptomatiktir. Makula veya optik diski etkilediğinde görme bozukluğuna neden olur.
SWS: Konjenital glokom eşlik ettiğinde kornea bulanıklığı, sulanma ve fotofobi oluşur. Geç başlangıçlı glokomda ağrısız ilerleyici görme alanı defekti meydana gelir.
VHL: Erken evrede asemptomatiktir. Retinal kapiller hemanjiyom ilerledikçe eksüdatif değişiklikler, makula ödemi ve halka şeklinde beyaz lekeler ortaya çıkar ve görme azalmasına yol açar.
AT: Görme genellikle korunur. Göz hareket bozuklukları (nistagmus, oküler motor apraksi) baskındır.
Lisch nodülleri: NF1’de en sık görülen göz bulgusudur. İriste çok sayıda, açık kahverengi, keskin sınırlı, kubbe şeklinde küçük nodüller (1-2 mm’den küçük). Yaşa göre prevalans: 3 yaş altı %5, 3-4 yaş %42, 5-6 yaş %55, 21 yaş üstü %100 olup yaşla artar. NF1 tanı kriterlerinde (2 veya daha fazla) yer alır. Japonlarda iris rengi kahverengi olduğundan yarık lamba ile dikkatli muayene önemlidir.
Optik yol tümörleri (optik gliom): NF1 hastalarının yaklaşık %15-25’inde görülür. Çoğunlukla düşük dereceli pilositik astrositomdur ve sıklıkla asemptomatiktir. İlerlediğinde optik atrofi, görme azalması ve görme alanı defektlerine neden olur. Kiazma tutulumu da olabilir.
Pleksiform nörofibrom: NF1 hastalarının %10’undan azında görülür. Göz kapağında “S şeklinde deformite” ile karakterizedir ve palpasyonda “kurt torbası” hissi verir. Eksoftalmi, şaşılık, ambliyopi ve konjenital glokoma neden olabilir.
Sfenoid kanat displazisi: Yörünge duvarının konjenital defekti olup pulsatil ekzoftalmiye yol açabilir.
Glokom: NF1’in %1-2’sinde görülür. Konjenital (tek taraflı) ve geç başlangıçlı olmak üzere iki tipi vardır.
Diğerleri: Korneada belirgin miyelinli sinirler, koroid kalınlaşması, retinal miyelinli sinirler.
Ekspoze keratit: Bilateral akustik nöroma nedeniyle 5. ve 7. kraniyal sinir hasarı yüz uyuşması, diplopi ve lagoftalmiye yol açar ve buna sekonder gelişir.
Retinal astrositik hamartom: Yaklaşık %50’sinde eşlik eder. Üç tipe ayrılır: (i) düz, yarı saydam, nonkalsifiye tip, (ii) kabarık, multinodüler, kalsifiye tip (dut görünümü), (iii) geçiş tipi. Genellikle arka kutupta birden fazla görülür.
Üç ana bulgu: (1) trigeminal sinir alanında yüz hemanjiyomu, (2) aynı tarafta intrakraniyal hemanjiyom, (3) aynı tarafta glokom veya koroidal hemanjiyomdur.
Glokom: SWS’de en önemli göz bulgusu. %30-70 oranında görülür. Konjenital glokom (doğumdan 4 yaşına kadar) yaklaşık %60’ını oluşturur ve buftalmus, kornea bulanıklığı, megakorneaya yol açar. Etiyolojide açı gelişim anomalisi, episkleral venöz basınç artışı ve koroidal hemanjiyomun rol oynadığı düşünülmektedir. Göz kapağı hemanjiyomu varlığında daha sık görülür.
Koroid hemanjiomu: Yaklaşık %20-70 oranında görülür. Yaygın ve sınırları belirsiz olduğu için normal fundus muayenesinde tanımlanması zordur. Fundus “ketçap benzeri” bir görünüm alır. Genellikle büyüme eğilimi göstermez, ancak eksüdatif değişiklikler ve eksüdatif retina dekolmanına yol açabilir.
Diğerleri: Konjonktiva, episklera ve iriste vasküler dilatasyon ve tortuozite.
Retinal kapiller hemanjiom (hemanjioblastom): VHL hastalarının %43-85’inde (ülkemiz raporlarında yaklaşık %60) görülür. Yaklaşık üçte biri bilateral ve multipltir. Retinanın temporal periferinde sık görülür ve dilate, tortuozite gösteren afferent ve efferent damarlarla birlikte kırmızı-turuncu nodül olarak izlenir. Ortalama başlangıç yaşı 25 olup genellikle 30 yaşına kadar ortaya çıkar.
İlerledikçe eksüdatif değişiklikler, retina kanaması, makula ödemi, halka şeklinde eksüda ve görme azalmasına yol açar.
Daha da ilerlerse vitreus kanaması, traksiyonel retina dekolmanı, neovasküler glokom ve sonunda körlüğe neden olabilir.
Floresein anjiyografide, besleyici arterden drene edici vene boya akışı görülür ve tümör bölgesinde erken dönemde belirgin boya sızıntısı izlenir.
Bulber konjonktiva telanjiektazisi: En sık görülen göz bulgusu. Genellikle 5-8 yaşlarında ortaya çıkar ve hastaların %80-90’ında görülür1).
Göz hareket bozuklukları: Nistagmus, okülomotor apraksi, sakkad anormallikleri, konverjans ve akomodasyon anormallikleri ile şaşılık görülür.
Görme genellikle korunur.
QLisch nodülleri yaşla birlikte nasıl değişir?
A
NF1 ile ilişkili Lisch nodüllerinin görülme sıklığı yaşla birlikte artar. 3 yaş altında %5 iken, 21 yaş üstünde neredeyse %100 oranında görülür. Genç yaşlarda yarık lamba muayenesinde bile tespit edilmesi zor olabilir ve yorum yaş dikkate alınarak yapılmalıdır.
Her hastalığın genetik özellikleri aşağıdaki gibidir.
NF1: Yaklaşık %50’si de novo mutasyondur. Penetrans neredeyse tamdır ancak fenotip çeşitlidir. Nörofibromin, RAS-GAP olarak işlev görür ve GTP-RAS’ı GDP-RAS’a dönüştürür2).
NF2: Merlin proteininin işlev kaybı. Schwann hücreleri ve leptomeningeal hücrelerde ağırlıklı olarak eksprese edilen bir tümör baskılayıcıdır; eksikliğinde schwannom, menenjiyom ve ependimom oluşur.
TSC: Sporadik vakalar yaklaşık üçte ikisini oluşturur. TSC2 mutasyonu sporadik vakaların %75-80’ini oluşturur ve TSC1 mutasyonundan daha şiddetli bir fenotipe sahiptir1). Hamartin/tüberin, mTOR yolunu doğrudan inhibe eder2).
SWS: Sporadiktir ve GNAQ genindeki somatik mozaik mutasyondan kaynaklanır1). Trigeminal sinirin birinci dalı (V1) bölgesinin tamamını kaplayan porto şarabı lekesi (PWB), oküler ve nörolojik komplikasyon riskini artırır.
VHL: Penetrans neredeyse %100’dür ve yaklaşık %20’si de novo mutasyondur. pVHL, HIF-α’nın ubikitinasyonu ve proteazomal yıkımında rol oynar2). Tip 1 (feokromositoma yok) yaklaşık %80, Tip 2 (feokromositoma var) yaklaşık %20’dir2).
AT: Otozomal resesif geçişli olup ATM kinaz, DNA çift sarmal kırıklarının onarımı ve genom stabilitesinin korunmasında rol oynar 1). Malign tümör (lenfatik) riski ve radyasyon duyarlılığı belirgin şekilde yüksektir.
NF1 tanı kriterleri (NIH kriterleri ve Japon Dermatoloji Derneği 2008) aşağıdaki 7 maddeden en az 2’sinin karşılanmasıyla kesin tanı konulmasını gerektirir.
6 veya daha fazla café-au-lait lekesi (ergenlik öncesi: en büyük çap ≥5 mm, ergenlik sonrası: ≥15 mm)
2 veya daha fazla nörofibrom veya diffüz nörofibrom
Koltuk altı ve kasık bölgesinde çillenme benzeri pigment lekeleri
Klinik kriterlerde bilateral vestibüler schwannom karakteristik bulgudur. Deri bulguları az olduğu için tanı NF1’den daha zor olabilir. Genetik testlerde %90’ın üzerinde mutasyon saptanır. Bilateral işitme siniri tümörleri BT ve MRG ile doğrulanır.
Kesin tanı, iki ana bulgu veya bir ana bulguya en az iki yardımcı bulgunun eşlik etmesiyle konur. TSC1/TSC2 patojenik varyantlarının saptanması bağımsız bir tanı kriteri olarak kabul edilir 1).
Retinal astrositik hamartomun değerlendirilmesinde fundus muayenesi, floresein anjiyografi ve OCT kullanılır. En önemli ayırıcı tanı retinoblastomdur. Çocukluk döneminde kalsifikasyon olmaması, besleyici damarların az olması ve sistemik semptomların eşlik etmesi ile ayırt edilebilir, ancak dut benzeri tümörden ayırt edilmesi bazen zordur.
Roach tanı skalasında, yüzde porto şarabı lekesi, göz içi basınç artışı ve leptomeningeal anjiyomdan en az ikisinin bulunması kriter olarak kabul edilir 1). Koroid anjiyomunun değerlendirilmesinde ED-OCT ve MRI yararlıdır. Kafa BT’sinde serebral kortekste kalsifikasyon görülür.
Glokom takibi için düzenli olarak göz içi basıncı ölçümü, optik sinir değerlendirmesi ve görme alanı testi yapılır. Bebeklerde yarık lamba muayenesine ek olarak anestezi altında fundus muayenesi ve göz içi basıncı ölçümü gerekebilir.
Genetik test, VHL mutasyonlarını neredeyse %100 oranında tespit edebilir ve günümüzde kesin tanının ana yöntemidir. Dilate fundus muayenesi 1 yaşından itibaren yılda bir kez önerilir. Periferde turuncu-kırmızı küresel tümörler ve genişlemiş kıvrımlı damarlar karakteristik bulgulardır. Floresein anjiyografi, küçük periferik hemanjiyoblastomların saptanmasında faydalıdır ve OCT, küçük lezyonların değerlendirilmesinde kullanılır.
Klinik tanıda ataksi, konjonktival telenjiektazi ve oküler motor anormallik üçlüsü önemlidir. Serum AFP yüksekliği, CA125 yüksekliği ve ATM protein eksikliği (Western blot) laboratuvar bulguları olarak görülür. Konjonktival telenjiektazi bu hastalık için patognomonik bir bulgudur. MRG, posterior fossada diffüz serebellar atrofi (özellikle vermis ve hemisferler) gösterir1).
Lisch nodülleri: Asemptomatik hamartomlardır ve tedavi gerektirmez.
Optik gliom: İlerleyici vakalarda cerrahi rezeksiyon düşünülür, ancak görme fonksiyonu kaybolur ve postoperatif komplikasyonlar fazladır. Kiazma invazyonu olan vakalarda kemoterapi endikedir.
Pleksiform nörofibrom: Tam cerrahi çıkarım zordur ve nüks sıktır. İlerleyici vakalarda orbital eksenterasyon gerekebilir. Rezeke edilemeyen vakalarda selumetinib (MEK inhibitörü) yeni tedavi olarak kullanılır1).
Glokom: Standart glokom tedavisine uygun yönetim yapılır.
Vestibüler schwannomda cerrahi rezeksiyon temeldir ve 3 cm’den küçük tümörlerde %65 vakada işitme korunabilir1). Ekspojur keratit suni gözyaşı, kornea koruyucu gözlük ve gerektiğinde tarsorafi ile tedavi edilir.
Retinal vasküler anormallikler (anevrizmal dilatasyon, arteriyovenöz malformasyon): Vitreus kanaması ve retina dekolmanı riski nedeniyle profilaktik fotokoagülasyon yapılır. Vitreus kanaması veya retina dekolmanı gelişirse vitrektomi düşünülür.
Epilepsi: İnfantil parsiyel nöbetler ve infantil spazmlarda vigabatrin birinci basamak tedavidir1). SEGA (subependimal dev hücreli astrositom) nöroşirürjikal rezeksiyon endikasyonudur1).
Glokom tedavisi başlangıç zamanına göre farklılık gösterir.
Konjenital glokom (erken başlangıçlı): Cerrahi tedavi zorunludur. Trabekülotomi ve gonyotomi tercih edilir. Koroid dekolmanı ve kanamaya karşı yeterli dikkat gereklidir; episkleral ven basıncı yüksekliği nedeniyle komplikasyon riski normalden yüksektir. İlaç tedavisine direnç genellikle yüksektir.
Geç başlangıçlı tip: Önce ilaç tedavisi uygulanır, etkisizse cerrahi düşünülür.
Koroid hemanjiomu tedavisi, eksüdatif değişikliklerin varlığına göre aşağıdakilerden seçilir.
Eksüdatif değişiklik oluşursa → fotokoagülasyon
Fotokoagülasyona yanıtsızlık veya eksüdatif retina dekolmanı → radyoterapi (toplam doz yaklaşık 20 Gray). Büllöz retina dekolmanının düzelmesi ve hemanjiomun küçülmesi beklenir.
Fotokoagülasyon (birinci seçenek): Argon veya diyot lazer kullanılarak, hemanjiyom çevresindeki retina ve besleyici damarlar yoğun şekilde koagüle edildikten sonra, tümörün kendisi doğrudan koagüle edilir. 2 disk çapından büyük tümörlerde, önce afferent arter ve çevre retina koagüle edilir, ardından tümör doğrudan koagüle edilir.
Kriyokoagülasyon: Fundus periferinde veya büyük hemanjiyomlarda fotokoagülasyonun zor olduğu durumlarda transskleral olarak uygulanır.
Vitrektomi: Eksüdatif değişikliklerin şiddetli olduğu, retina dekolmanı veya proliferatif değişikliklerin geliştiği durumlarda düşünülür.
İleri evre: Neovasküler glokomun yönetimi gerekir.
Aile öyküsü varsa tüm aile bireylerine fundus muayenesi yapılmalı ve erken tedavi ile iyi prognoz beklenebilir.
Konjonktival telenjiektaziler için spesifik bir tedavi yoktur. İmmün yetmezlik için profilaktik antibiyotikler ve intravenöz immünoglobulin (IVIg) uygulanır1). Ataksi tedavisi semptomatiktir.
QVHL'de retinal hemanjiyom için ne zaman tarama yapılmalıdır?
A
VHL’de 1 yaşından itibaren yılda bir kez dilatasyonlu fundus muayenesi önerilir. VHL gen mutasyonu doğrulanmış hastaların tüm aile bireylerinin de fundus muayenesi yaptırması önemlidir; erken tedavi ile iyi bir görsel prognoz beklenebilir.
Her hastalığın moleküler düzeydeki mekanizması aşağıda verilmiştir.
NF1 ve TSC (mTOR Yolağı)
NF1 (Nörofibromin): RAS-GAP olarak işlev görür. GTP bağlı RAS’ı (aktif) GDP bağlı RAS’a (inaktif) dönüştürmeyi hızlandırır. Mutasyon → RAS’ın sürekli aktivasyonu → MAP kinaz yolağı ve PI3K-Akt-mTOR yolağının düzensizliği → kontrolsüz hücre çoğalması 2).
TSC (Hamartin/Tüberin): mTOR kompleks 1 ve 2’yi doğrudan inhibe eden bir tümör baskılayıcı komplekstir. Mutasyon → mTOR aşırı aktivasyonu → enerji metabolizması, protein/lipid sentezi ve hücre sağkalımında anormallikler 2). TSC2 mutasyonu, TSC1 mutasyonuna göre daha şiddetli bir fenotipe yol açar 1).
VHL (HIF yolağı)
pVHL: E3 ubikitin ligaz kompleksinin (elongin B/C, Cullin 2, RBX1) bir bileşenidir. Normal oksijen koşullarında, prolil hidroksilaz HIF-α’yı hidroksile eder → pVHL tanır → ubikitinlenir → proteazomda yıkılır.
VHL mutasyonu: Sürekli olarak psödohipoksik durum oluşur → HIF-α birikir → HIF-1β ile heterodimer oluşur → VEGF, eritropoietin, metabolizma ve hücre çoğalması ile ilgili genlerin transkripsiyonu aktive olur → vasküler tümör oluşumu2).
NF2 (merlin): Schwann hücreleri ve leptomeningeal hücrelerde eksprese edilen tümör baskılayıcı protein. Mutasyonu schwannom, menenjiyom ve ependimom oluşumuna yol açar.
SWS (GNAQ somatik mozaik mutasyonu): G proteini ile ilişkili transmembran sinyal iletiminde anormallik. Embriyodaki mutasyonun zamanı ve yeri, trigeminal sinir alanı, kafa içi ve gözdeki ekspresyon paternini farklılaştırır1). GNAQ ayrıca SWS, KTS ve PPV’nin ortak moleküler temelidir3).
AT (ATM kinaz): DNA çift sarmal kırıklarının onarımı ve genomik kararlılığın korunmasında rol oynayan tümör baskılayıcı faktör. Mutasyon → DNA onarım bozukluğu → kanser yatkınlığı, radyasyon duyarlılığında belirgin artış ve immün yetmezlik1).
Chevalier ve ark. (2021) fokomatozlar ile endokrin tümörler arasındaki ilişkiyi incelemiş ve NF1, TSC ve VHL sinyal yollarının (RAS-PI3K-Akt-mTOR yolu, HIF yolu) multipl endokrin tümör gelişiminde ortak rol oynadığını bildirmiştir2).
QFokomatozlar neden birden çok organda lezyonlara neden olur?
A
Fokomatozların ortak patofizyolojik temeli, nöral krest hücrelerinin oluşumu, göçü ve farklılaşmasındaki anormalliktir. Nöral krest hücreleri ektoderm kaynaklıdır ve Schwann hücreleri, melanositler gibi çeşitli hücreleri üretir; bu nedenle sinir, deri ve göz gibi birden çok organda lezyonlar görülür. Ayrıca RAS-mTOR yolu ve VHL-HIF yolu gibi ortak sinyal yollarındaki mutasyonlar, organlar arası tümör oluşumunu teşvik eder.
7. Güncel araştırmalar ve geleceğe bakış (araştırma aşamasındaki raporlar)
VHL ile ilişkili berrak hücreli renal hücreli karsinom ve pankreatik nöroendokrin tümörler için Faz 2 çalışması devam etmektedir.
Chevalier ve ark. (2021) tarafından yapılan MK6482 çalışmasında, pankreatik nöroendokrin tümörlerin %64’ünde objektif yanıt elde edilmiş ve 12 aylık progresyonsuz sağkalım oranı %98.3 olarak bildirilmiştir2).
mTOR inhibitörü everolimus (TSC ile ilişkili tümörler)
TSC ile ilişkili renal anjiyomiyolipom ve SEGA için EXIST çalışmasında morfolojik yanıt doğrulanmış olup, endokrin tümörlerde de uygulanması beklenmektedir2).
GNAQ gen mutasyonunun SWS, Klippel-Trenaunay sendromu (KTS) ve pigmenter vasküler fakomatoz (PPV) için ortak bir moleküler temel olduğu bulunmuştur3). Bu örtüşen vakalarda koroidal melanom riski nedeniyle yılda 1-2 kez dilatasyonlu fundus muayenesi önerilmektedir3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.