Le facomatosi (phakomatoses) sono un gruppo di malattie congenite caratterizzate da lesioni amartomatose della pelle, del sistema nervoso centrale e degli occhi. Sono anche chiamate sindromi neurocutanee.
La denominazione è dovuta all’oftalmologo olandese Van der Hoeve, dal greco «phakos» (lente/macchia). Inizialmente includeva tre malattie: neurofibromatosi, sclerosi tuberosa e malattia di von Hippel-Lindau, successivamente sono state aggiunte la sindrome di Sturge-Weber e l’atassia telangiectasica. Attualmente sono descritte oltre 60 sindromi.
La base patogenetica comune è un’anomalia della formazione, migrazione e differenziazione delle cellule della cresta neurale. Le cellule della cresta neurale derivano dall’ectoderma e producono diverse cellule come cellule di Schwann e melanociti, causando lesioni in più organi: nervi, pelle e occhi. Le vie di segnalazione coinvolte includono RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ e VHL-HIF.
Le frequenze di occorrenza delle sei principali malattie sono le seguenti.
Le sei malattie rappresentative sono NF1, NF2, sclerosi tuberosa, sindrome di Sturge-Weber, malattia di von Hippel-Lindau e atassia teleangectasica. Tutte sono causate da mutazioni genetiche e provocano lesioni in più organi, inclusi i sistemi nervoso, cutaneo e oculare. Attualmente, oltre 60 sindromi sono incluse nella categoria delle facomatosi.
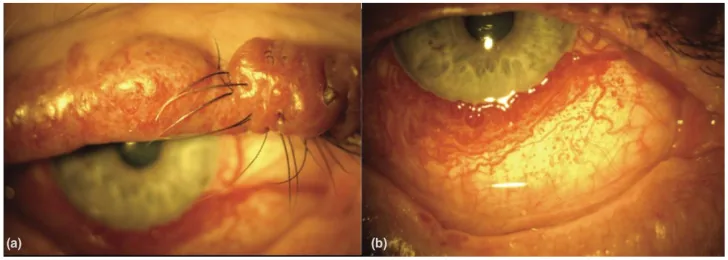
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Macchia color vino di Porto della palpebra superiore con nodularità in un paziente con sindrome di Sturge-Weber. (b) Vascolarizzazione congiuntivale diffusa in un paziente con sindrome di Sturge-Weber. Da [15].
I tipi di complicanze oculari differiscono per ciascuna malattia e anche i sintomi soggettivi sono vari.
NF1 : La progressione di un tumore della via ottica (glioma ottico) causa diminuzione dell’acuità visiva, perdita della visione dei colori e difetti del campo visivo. Il neurofibroma plessiforme può causare esoftalmo.
Sclerosi tuberosa : L’amartoma retinico a cellule astrocitiche è solitamente asintomatico. Se colpisce la macula o la papilla ottica, causa deficit visivo.
SWS : In caso di glaucoma congenito, si verificano opacità corneale, lacrimazione e fotofobia. Nel glaucoma a esordio tardivo si verifica un difetto del campo visivo progressivo e indolore.
VHL: inizialmente asintomatico. Con la progressione dell’emangioma capillare retinico compaiono alterazioni essudative, edema maculare e macchie bianche anulari, con conseguente calo visivo.
AT: l’acuità visiva è generalmente conservata. Predominano i disturbi della motilità oculare (nistagmo, aprassia oculomotoria).
Noduli di Lisch: il reperto oculare più frequente nella NF1. Piccoli noduli (meno di 1–2 mm) di colore marrone chiaro, a bordi netti, a forma di cupola, multipli sull’iride. La prevalenza per età aumenta con l’età: <3 anni 5%, 3–4 anni 42%, 5–6 anni 55%, ≥21 anni 100%; è inclusa nei criteri diagnostici della NF1 (≥2). Nei giapponesi, a causa del colore bruno dell’iride, è importante un esame accurato con lampada a fessura.
Tumore delle vie ottiche (glioma ottico): si verifica in circa il 15–25% dei pazienti con NF1. Per lo più astrocitoma pilocitico di basso grado, spesso asintomatico. Con la progressione può causare atrofia ottica, calo visivo e difetti del campo visivo. Possibile infiltrazione chiasmatica.
Neurofibroma plessiforme: si verifica in meno del 10% dei pazienti con NF1. Caratterizzato da una «deformità a S» della palpebra, con una sensazione di «sacco di vermi» alla palpazione. Può causare esoftalmo, strabismo, ambliopia e glaucoma congenito.
Displasia dell’ala dello sfenoide: difetto congenito della parete orbitaria che può causare esoftalmo pulsante.
Glaucoma: si verifica nell’1-2% dei pazienti con NF1. Esistono due tipi: congenito (unilaterale) e a esordio tardivo.
Cheratite da esposizione: secondaria a neurinoma bilaterale dell’acustico che causa danno ai nervi cranici V e VII, provocando intorpidimento facciale, diplopia e lagoftalmo.
I noduli di Lisch sono rari.
Manifestazioni oculari della sclerosi tuberosa (TSC)
Amartoma astrocitico retinico: presente in circa il 50% dei casi. Classificato in 3 tipi: (i) piatto, traslucido, non calcificato; (ii) rilevato, multinodulare, calcificato (aspetto a «gelso»); (iii) tipo di transizione. Di solito multipli al polo posteriore.
La triade classica comprende (1) angioma facciale nel territorio del trigemino, (2) angioma intracranico omolaterale, (3) glaucoma o angioma coroideo omolaterale.
Glaucoma : il segno oculare più importante nella SWS, presente nel 30-70% dei casi. Il glaucoma congenito (dalla nascita ai 4 anni) rappresenta circa il 60% dei casi, causando buftalmo, opacità corneale e megalocornea. L’eziologia include displasia dell’angolo, aumento della pressione venosa episclerale e coinvolgimento di un angioma coroideo. Si verifica frequentemente in presenza di angioma palpebrale.
Emangioma coroideo: si verifica in circa il 20-70% dei casi. È diffuso e mal definito, quindi difficile da identificare con un esame del fondo oculare normale. Il fondo oculare ha un aspetto a «ketchup di pomodoro». Di solito non mostra tendenza all’aumento, ma può causare alterazioni essudative e distacco di retina essudativo.
Altro : dilatazione e tortuosità dei vasi della congiuntiva, dell’episclera e dell’iride.
Segni oculari della malattia di VHL (von Hippel-Lindau)
Emangioma capillare retinico (emangioblastoma): si verifica nel 43-85% dei pazienti con VHL (circa il 60% nei rapporti nazionali). Circa un terzo dei casi è bilaterale e multiplo. Predilige la periferia temporale della retina e si osserva come un nodulo rosso-arancio con vasi afferenti ed efferenti dilatati e tortuosi. L’età media di insorgenza è di 25 anni, di solito compare entro i 30 anni.
Con il progredire, si verificano alterazioni essudative, emorragia retinica, edema maculare, macchie bianche anulari e diminuzione dell’acuità visiva.
L’angiografia con fluoresceina mostra un flusso di colorante dall’arteria afferente alla vena efferente, con una precoce e marcata fuoriuscita di colorante a livello della massa tumorale.
Telangectasie congiuntivali : il segno oculare più comune. Di solito si manifestano entro i 5-8 anni e sono presenti nell’80-90% dei pazienti1).
Disturbi dei movimenti oculari : nistagmo, aprassia oculomotoria, anomalie delle saccadi, anomalie della convergenza e dell’accomodazione, strabismo.
L’acuità visiva è generalmente conservata.
QCome cambiano i noduli di Lisch con l'età?
A
I noduli di Lisch associati alla NF1 aumentano di frequenza con l’età. Nei bambini di età inferiore a 3 anni sono presenti solo nel 5% dei casi, ma in quelli di età superiore a 21 anni si riscontrano in quasi il 100%. Nei pazienti giovani, possono essere difficili da rilevare anche all’esame con lampada a fessura, pertanto è necessaria un’interpretazione che tenga conto dell’età.
Le caratteristiche genetiche di ciascuna malattia sono le seguenti.
NF1: circa il 50% sono mutazioni de novo. Penetranza quasi completa ma fenotipo variabile. La neurofibromina funge da RAS-GAP, convertendo GTP-RAS in GDP-RAS2).
NF2: perdita di funzione della proteina merlina. Soppressore tumorale espresso principalmente nelle cellule di Schwann e nelle cellule meningee; la sua carenza porta a schwannomi, meningiomi ed ependimomi.
TSC: circa 2/3 dei casi sono sporadici. Le mutazioni TSC2 rappresentano il 75-80% dei casi sporadici e hanno un fenotipo più grave rispetto alle mutazioni TSC11). L’amartina/tuberina inibisce direttamente la via mTOR2).
SWS: sporadico, causato da mutazione somatica a mosaico del gene GNAQ1). Una macchia color vino (PWB) che copre l’intera area del primo ramo del trigemino (V1) comporta un alto rischio di complicanze oftalmologiche e neurologiche.
VHL: penetranza quasi al 100%, circa il 20% di mutazioni de novo. pVHL è coinvolto nell’ubiquitinazione e degradazione proteasomica di HIF-α2). Tipo 1 (senza feocromocitoma) circa l’80%, Tipo 2 (con feocromocitoma) circa il 20%2).
AT : malattia autosomica recessiva in cui la chinasi ATM è coinvolta nella riparazione delle rotture del doppio filamento del DNA e nel mantenimento della stabilità genomica 1). Il rischio di tumori maligni (linfoidi) e la radiosensibilità sono notevolmente elevati.
I criteri diagnostici per la NF1 (criteri NIH, Società Giapponese di Dermatologia 2008) stabiliscono che la diagnosi definitiva viene posta quando sono soddisfatti almeno 2 dei seguenti 7 criteri.
6 o più macchie caffè-latte (prima della pubertà: diametro massimo ≥5 mm; dopo la pubertà: ≥15 mm)
2 o più neurofibromi o un neurofibroma plessiforme
Lesioni ossee caratteristiche (ad esempio, displasia dell’ala dello sfenoide)
Un parente di primo grado affetto dalla stessa malattia
Il ruolo dell’esame oftalmologico è il seguente.
Lampada a fessura: Rilevamento dei noduli di Lisch (a volte difficile nei giovani).
OCT del segmento anteriore e microscopia ultrasonica: Utili per il rilevamento ausiliario dei noduli di Lisch.
RM: Miglior metodo diagnostico per i tumori delle vie ottiche. Essenziale per la valutazione del chiasma e del tratto ottico.
Esame del campo visivo: Screening per tumori delle vie ottiche (difficile da eseguire nei bambini piccoli).
Tomografia a coerenza ottica (OCT) : valutazione dell’assottigliamento dello strato delle fibre nervose retiniche associato a tumori delle vie ottiche.
PEV (potenziali evocati visivi) : rilevamento precoce del danno al nervo ottico.
Intervallo di controllo regolare : solo noduli di Lisch → 1 volta all’anno. In presenza di glioma ottico → 1 volta ogni 3 mesi.
I criteri clinici sono caratterizzati dallo schwannoma vestibolare bilaterale. La diagnosi può essere più difficile rispetto alla NF1 a causa della scarsità di sintomi cutanei. I test genetici rilevano oltre il 90% delle mutazioni. La TC e la RM confermano i neurinomi acustici bilaterali.
La diagnosi è confermata da 2 criteri maggiori, o 1 criterio maggiore + 2 o più criteri minori. L’identificazione di una mutazione patogenetica di TSC1/TSC2 è riconosciuta come criterio diagnostico indipendente 1).
La valutazione dell’amartoma retinico a cellule astrocitiche utilizza l’esame del fondo oculare, l’angiografia con fluoresceina e l’OCT. La diagnosi differenziale più importante è il retinoblastoma. La differenziazione è possibile per l’assenza di calcificazione, la scarsità di vasi nutritizi e la presenza di sintomi sistemici in età pediatrica, ma la distinzione dal tumore a mora può essere difficile.
Secondo la scala diagnostica di Roach, la diagnosi richiede almeno due dei seguenti: macchia color vino porto facciale, aumento della pressione intraoculare e angioma leptomeningeo 1). L’ED-OCT e la RM sono utili per la valutazione dell’angioma coroideale. La TC cranica mostra calcificazioni nella corteccia cerebrale.
Il monitoraggio del glaucoma include misurazioni regolari della pressione intraoculare, valutazione del nervo ottico ed esami del campo visivo. Nei neonati, oltre all’esame con lampada a fessura, possono essere necessari esame del fondo oculare e misurazione della pressione intraoculare in anestesia.
I test genetici rilevano mutazioni VHL in quasi il 100% dei casi e sono attualmente il metodo principale per la diagnosi definitiva. Si raccomanda un esame del fondo oculare con dilatazione pupillare una volta all’anno a partire da 1 anno di età. I tumori sferici rosso-arancio periferici con vasi tortuosi dilatati sono reperti caratteristici. L’angiografia con fluoresceina è utile per rilevare piccoli emangioblastomi periferici e l’OCT viene utilizzata per valutare piccole lesioni.
Per la diagnosi clinica è importante la triade: atassia, teleangectasie congiuntivali e anomalie dei movimenti oculari. I reperti di laboratorio includono aumento dell’AFP sierica, aumento del CA125 e deficit della proteina ATM (Western blot). Le teleangectasie congiuntivali sono patognomoniche per questa malattia. La RM mostra atrofia cerebellare diffusa della fossa cranica posteriore (in particolare del verme e degli emisferi)1).
Noduli di Lisch: amartomi asintomatici che non richiedono trattamento.
Glioma ottico : in caso di progressione si considera la resezione chirurgica, ma la funzione visiva è persa e le complicanze postoperatorie sono frequenti. In caso di infiltrazione chiasmatica è indicata la chemioterapia.
Neurofibroma plessiforme : l’asportazione chirurgica totale è difficile e le recidive sono frequenti. Nei casi progressivi può essere necessaria l’exenteratio orbitae. Nei casi non resecabili, il selumetinib (inibitore di MEK) viene utilizzato come nuovo trattamento 1).
Glaucoma : gestione secondo il trattamento standard del glaucoma.
Per lo schwannoma vestibolare, la resezione chirurgica è il trattamento di base; per tumori inferiori a 3 cm, la conservazione dell’udito è possibile nel 65% dei casi 1). La cheratite da esposizione viene trattata con lacrime artificiali, occhiali protettivi corneali e, se necessario, tarsorrafia.
Epilessia: La vigabatrina è considerata il trattamento di prima linea per le crisi parziali e gli spasmi infantili (sindrome di West) nei neonati1). Il SEGA (astrocitoma subependimale a cellule giganti) è un’indicazione per la resezione neurochirurgica1).
Il trattamento del glaucoma varia a seconda dell’età di insorgenza.
Glaucoma congenito (esordio precoce) : Il trattamento chirurgico è obbligatorio. Si scelgono trabeculotomia e goniotomia. È necessaria un’attenzione adeguata al distacco coroidale e all’emorragia, poiché il rischio di complicanze è più elevato del normale a causa dell’aumento della pressione venosa episclerale. La resistenza al trattamento farmacologico tende ad essere elevata.
Forma tardiva : prima trattamento farmacologico, se inefficace, considerare l’intervento chirurgico.
Il trattamento dell’angioma coroideale viene scelto in base alla presenza o meno di alterazioni essudative tra le seguenti opzioni.
In caso di alterazioni essudative → fotocoagulazione
Fotocoagulazione inefficace o distacco di retina essudativo → radioterapia (dose totale di circa 20 gray). Ci si può aspettare il riattacco del distacco di retina bolloso e la riduzione dell’angioma.
Fotocoagulazione (prima scelta) : Utilizzo di laser argon o dye, coagulazione densa della retina intorno all’emangioma e dei vasi nutritizi, quindi coagulazione diretta del tumore. Per tumori di diametro superiore a 2 diametri papillari, coagulare l’arteria afferente e la retina circostante prima di coagulare direttamente il tumore.
Criocoagulazione : Eseguita per via transclerale quando la fotocoagulazione è difficile a causa della localizzazione periferica del fondo o di un emangioma gigante.
Vitrectomia : considerata in caso di alterazioni essudative gravi, distacco di retina o alterazioni proliferative.
In caso di storia familiare, eseguire un esame del fondo oculare per tutti i membri della famiglia; un trattamento precoce può portare a una buona prognosi.
Non esiste un trattamento specifico per le teleangectasie congiuntivali. Per l’immunodeficienza vengono somministrati antibiotici profilattici e immunoglobuline per via endovenosa (IVIg) 1). L’atassia viene trattata principalmente con terapia sintomatica.
QQuando iniziare lo screening per gli emangiomi retinici nella malattia di VHL?
A
Nella VHL si raccomanda un esame del fondo oculare annuale con dilatazione pupillare a partire da 1 anno di età. È importante che anche tutti i familiari dei pazienti con mutazione confermata del gene VHL si sottopongano all’esame del fondo oculare, poiché un trattamento precoce può portare a una buona prognosi visiva.
I meccanismi molecolari di ciascuna malattia sono presentati di seguito.
NF1 e TSC (via mTOR)
NF1 (neurofibromina) : funge da RAS-GAP. Promuove la conversione di RAS legato a GTP (forma attiva) in RAS legato a GDP (forma inattiva). La mutazione porta a un’attivazione persistente di RAS, alla deregolazione delle vie MAP chinasi e PI3K-Akt-mTOR e a una proliferazione cellulare incontrollata2).
TSC (amartina/tuberina) : complesso soppressore tumorale che inibisce direttamente i complessi mTOR 1 e 2. La mutazione porta a iperattivazione di mTOR, anomalie del metabolismo energetico, della sintesi proteica/lipidica e della sopravvivenza cellulare2). Le mutazioni di TSC2 causano un fenotipo più grave rispetto alle mutazioni di TSC11).
VHL (via HIF)
pVHL : componente del complesso E3 ubiquitina ligasi (elongin B/C, Cullin 2, RBX1). In normossia, la prolil idrossilasi idrossila HIF-α → pVHL lo riconosce → ubiquitinazione → degradazione proteasomica.
Mutazione VHL : si verifica costantemente uno stato pseudo-ipossico → accumulo di HIF-α → formazione di eterodimeri con HIF-1β → attivazione trascrizionale dei geni correlati a VEGF, produzione di globuli rossi, metabolismo e proliferazione cellulare → formazione di tumori vascolari2).
NF2 (merlina) : proteina oncosoppressore espressa nelle cellule di Schwann e nelle cellule leptomeningee. Le mutazioni causano schwannomi, meningiomi ed ependimomi.
SWS (mutazione somatica a mosaico di GNAQ) : anomalia della segnalazione transmembrana associata alle proteine G. Il momento e il luogo della mutazione nell’embrione determinano i modelli di espressione nel territorio del nervo trigemino, intracranico e nell’occhio1). GNAQ è anche la base molecolare comune per SWS, KTS e PPV3).
AT (chinasi ATM) : soppressore tumorale coinvolto nella riparazione delle rotture del doppio filamento del DNA e nel mantenimento della stabilità genomica. Mutazione → alterazione della riparazione del DNA → predisposizione al cancro, marcato aumento della radiosensibilità, immunodeficienza1).
Chevalier et al. (2021) hanno esaminato l’associazione tra facomatosi e tumori endocrini e hanno riportato che le vie di segnalazione di NF1, TSC e VHL (via RAS-PI3K-Akt-mTOR, via HIF) sono coinvolte in comune nello sviluppo di tumori endocrini multipli 2).
QPerché le facomatosi causano lesioni in più organi?
A
La base fisiopatologica comune delle facomatosi è un’anomalia nella formazione, migrazione e differenziazione delle cellule della cresta neurale. Le cellule della cresta neurale derivano dall’ectoderma e producono diverse cellule come cellule di Schwann e melanociti, portando a lesioni in più organi: nervoso, cutaneo e oculare. Inoltre, le mutazioni in vie di segnalazione comuni come la via RAS-mTOR e la via VHL-HIF promuovono lo sviluppo di tumori oltre i confini degli organi.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
È in corso uno studio di fase 2 per il carcinoma renale a cellule chiare e i tumori neuroendocrini pancreatici associati a VHL.
Lo studio di MK6482 condotto da Chevalier et al. (2021) ha riportato un tasso di risposta obiettiva del 64% nei tumori neuroendocrini pancreatici, con un tasso di sopravvivenza libera da progressione a 12 mesi del 98,3% 2).
Inibitore mTOR everolimus (tumori associati a TSC)
Lo studio EXIST per angiomiolipoma renale e SEGA associati a TSC ha confermato una risposta morfologica, e si prevede anche un’applicazione ai tumori endocrini 2).
È stato scoperto che la mutazione del gene GNAQ è una base molecolare comune per la sindrome di Sturge-Weber (SWS), la sindrome di Klippel-Trénaunay (KTS) e la facomatosi pigmentovascolare (PPV)3). A causa del rischio di melanoma coroidale in questi casi di sovrapposizione, si raccomanda un esame del fondo oculare in midriasi 1-2 volte l’anno3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.