Las facomatosis son un grupo de trastornos congénitos caracterizados por lesiones hamartomatosas en la piel, el sistema nervioso central y los ojos. También se denominan síndromes neurocutáneos.
El término fue acuñado por el oftalmólogo holandés Van der Hoeve, derivado de la palabra griega “phakos” (lente o mancha). Inicialmente incluía la neurofibromatosis, la esclerosis tuberosa y la enfermedad de von Hippel-Lindau, luego se agregaron el síndrome de Sturge-Weber y la ataxia telangiectasia. Actualmente se han descrito más de 60 síndromes.
La base patológica común es una anomalía en la formación, migración y diferenciación de las células de la cresta neural. Dado que las células de la cresta neural se derivan del ectodermo y producen diversas células como células de Schwann y melanocitos, se producen lesiones en múltiples órganos, incluidos nervios, piel y ojos. Las vías de señalización involucradas incluyen RAS, MAPK/MEK, mTOR, PI3K/AKT, GNAQ y VHL-HIF.
La incidencia de las seis enfermedades principales es la siguiente.
Q¿Qué enfermedades se incluyen en las facomatosis?
A
Las seis enfermedades representativas son NF1, NF2, esclerosis tuberosa, síndrome de Sturge-Weber, enfermedad de von Hippel-Lindau y ataxia telangiectasia. Todas son causadas por mutaciones genéticas y producen lesiones en múltiples órganos, incluidos el sistema nervioso, la piel y los ojos. Actualmente, más de 60 síndromes se incluyen en la categoría de facomatosis.
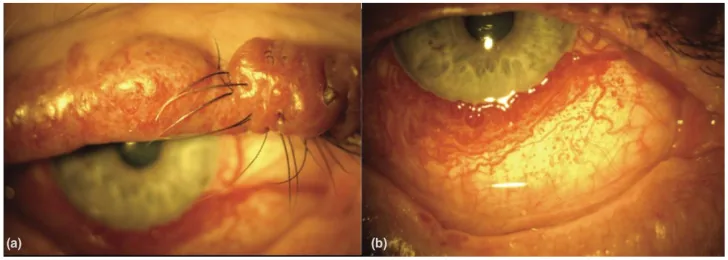
An Update on Multimodal Ophthalmological Imaging of Diffuse Choroidal Hemangioma in Sturge–Weber Syndrome. Vision (Basel). 2023 Oct 6; 7(4):64. Figure 1. PMCID: PMC10594527. License: CC BY.
(a) Mancha en vino de Oporto del párpado superior con nodularidad en un paciente con síndrome de Sturge-Weber. (b) Vascularización conjuntival difusa en un paciente con síndrome de Sturge-Weber. De [15].
Los tipos de complicaciones oculares varían según la enfermedad, y los síntomas subjetivos también son diversos.
NF1: A medida que progresan los tumores de la vía óptica (gliomas ópticos), se producen pérdida de agudeza visual, pérdida de visión cromática y defectos del campo visual. Los neurofibromas plexiformes pueden causar proptosis.
Esclerosis tuberosa: Los hamartomas astrocíticos retinianos suelen ser asintomáticos. La discapacidad visual ocurre cuando se afectan la mácula o el disco óptico.
SWS: Cuando se asocia con glaucoma congénito, se producen opacidad corneal, lagrimeo y fotofobia. El glaucoma de aparición tardía causa pérdida progresiva e indolora del campo visual.
VHL: Asintomático en etapas tempranas. A medida que el hemangioma capilar retiniano progresa, aparecen cambios exudativos, edema macular y retinopatía circinada, lo que lleva a disminución de la visión.
AT: La visión generalmente se conserva. Predominan los trastornos oculomotores (nistagmo, apraxia oculomotora).
Nódulos de Lisch: El hallazgo ocular más frecuente en NF1. Nódulos pequeños (<1–2 mm), de color marrón claro, bien definidos, en forma de cúpula, múltiples en el iris. La prevalencia por edad aumenta con la edad: <3 años 5%, 3–4 años 42%, 5–6 años 55%, ≥21 años 100%, y se incluye en los criterios diagnósticos de NF1 (≥2 nódulos). En individuos japoneses, el color del iris es marrón, por lo que es importante un examen cuidadoso con lámpara de hendidura.
Tumores de la vía óptica (gliomas ópticos): Ocurren en aproximadamente el 15–25% de los pacientes con NF1. La mayoría son astrocitomas pilocíticos de bajo grado y a menudo asintomáticos. La progresión puede provocar atrofia óptica, deterioro visual y defectos del campo visual. Puede haber afectación quiasmática.
Neurofibroma plexiforme: Ocurre en menos del 10% de los pacientes con NF1. Se caracteriza por una “deformidad en forma de S” del párpado, con una textura de “bolsa de gusanos” a la palpación. Puede causar proptosis, estrabismo, ambliopía y glaucoma congénito.
Displasia del ala esfenoidal: Defecto congénito de la pared ósea orbitaria, que puede causar proptosis pulsátil.
Glaucoma: Ocurre en el 1–2% de los pacientes con NF1. Hay dos tipos: congénito (unilateral) y de inicio tardío.
Queratitis por exposición: La disfunción de los nervios craneales V y VII debido a neuromas acústicos bilaterales causa entumecimiento facial, diplopía y lagoftalmos, lo que lleva a queratitis por exposición secundaria.
Los nódulos de Lisch son raros.
Hallazgos oculares en el complejo de esclerosis tuberosa (TSC)
Hamartoma astrocítico retiniano: Ocurre en aproximadamente el 50% de los casos. Se clasifica en tres tipos: (i) plano, translúcido, no calcificado; (ii) elevado, multinodular, calcificado (apariencia de mora); (iii) tipo de transición. Generalmente se observan múltiples lesiones en el polo posterior.
Lesiones retinianas despigmentadas: Aparecen como lesiones “en sacabocados”.
Anomalías vasculares retinianas: Pueden presentarse dilataciones aneurismáticas y malformaciones arteriovenosas, que pueden causar hemorragia vítrea, retinopatía vítrea proliferativa y desprendimiento de retina.
Otros: Angiofibromas palpebrales, manchas despigmentadas del iris, coloboma coroideo atípico.
Hallazgos oculares en el SWS (síndrome de Sturge-Weber)
Las tres características principales son (1) hemangioma facial en la distribución del nervio trigémino, (2) hemangioma intracraneal ipsilateral y (3) glaucoma ipsilateral o hemangioma coroideo.
Glaucoma: El hallazgo ocular más importante en el SWS. Ocurre en el 30–70% de los casos. El glaucoma congénito (desde el nacimiento hasta los 4 años) representa aproximadamente el 60%, causando buftalmos, opacidad corneal y megalocórnea. Se cree que la etiología involucra disgenesia angular, elevación de la presión venosa epiescleral y hemangioma coroideo. Ocurre con frecuencia en presencia de hemangioma palpebral.
Hemangioma coroideo: Ocurre en aproximadamente el 20-70% de los casos. Es difuso con bordes indistintos, lo que dificulta su identificación en el examen de fondo de ojo de rutina. El fondo de ojo tiene una apariencia de “salsa de tomate”. Por lo general, no muestra tendencia a agrandarse, pero puede causar cambios exudativos y desprendimiento de retina exudativo.
Otros: Dilatación y tortuosidad vascular de la conjuntiva, epiesclera e iris.
Hallazgos oculares en la enfermedad de VHL (von Hippel-Lindau)
Hemangioma capilar retiniano (hemangioblastoma): Ocurre en el 43-85% de los pacientes con VHL (aproximadamente el 60% en informes japoneses). Aproximadamente un tercio son bilaterales y múltiples. Se presenta comúnmente en la periferia temporal de la retina y se observa como un nódulo rojo anaranjado con vasos aferentes y eferentes dilatados y tortuosos. La edad media de inicio es de 25 años y generalmente aparece antes de los 30 años.
La progresión conduce a cambios exudativos, hemorragia retiniana, edema macular, retinopatía circinada y disminución de la agudeza visual.
La angiografía fluoresceínica muestra la entrada de colorante en la arteria aferente → perfusión de la vena eferente, con una fuga de colorante marcada desde la zona tumoral en la fase temprana.
Hallazgos oculares en la AT (ataxia telangiectasia)
Telangiectasia conjuntival bulbar: El hallazgo ocular más común. Generalmente aparece entre los 5 y 8 años y se observa en el 80–90% de los pacientes1).
Anomalías oculomotoras: Se observan nistagmo, apraxia oculomotora, sacadas anormales, anomalías de convergencia y acomodación, y estrabismo.
La agudeza visual generalmente se conserva.
Q¿Cómo cambian los nódulos de Lisch con la edad?
A
Los nódulos de Lisch asociados con NF1 aumentan en frecuencia con la edad. Están presentes solo en el 5% de los niños menores de 3 años, pero se encuentran en casi el 100% de las personas de 21 años o más. En niños pequeños, pueden ser difíciles de detectar incluso con examen con lámpara de hendidura, por lo que se requiere una interpretación ajustada por edad.
Las características genéticas de cada enfermedad son las siguientes.
NF1: Aproximadamente el 50% son mutaciones de novo. Penetrancia casi completa pero expresividad variable. La neurofibromina funciona como RAS-GAP, convirtiendo GTP-RAS en GDP-RAS2).
NF2: Pérdida de función de la proteína merlina. Supresor tumoral expresado principalmente en células de Schwann y células meníngeas; su deficiencia produce schwannomas, meningiomas y ependimomas.
TSC: Los casos esporádicos representan aproximadamente dos tercios. Las mutaciones de TSC2 representan el 75-80% de los casos esporádicos y producen un fenotipo más grave que las mutaciones de TSC11). La hamartina/tuberina inhibe directamente la vía mTOR2).
SWS: Esporádico, causado por mutaciones somáticas en mosaico del gen GNAQ1). La mancha color vino de Oporto (PWB) que afecta toda la primera rama (V1) del nervio trigémino conlleva un alto riesgo de complicaciones oculares y neurológicas.
VHL: La penetrancia es casi del 100%, con aproximadamente un 20% de mutaciones de novo. pVHL participa en la ubiquitinación y degradación proteasómica de HIF-α2). El tipo 1 (sin feocromocitoma) representa aproximadamente el 80%, y el tipo 2 (con feocromocitoma) aproximadamente el 20%2).
AT: Herencia autosómica recesiva; la quinasa ATM participa en la reparación de roturas de doble cadena de ADN y el mantenimiento de la estabilidad genómica 1). El riesgo de tumores malignos (linfoides) y la radiosensibilidad son notablemente altos.
Los criterios diagnósticos para NF1 (criterios NIH, Sociedad Japonesa de Dermatología 2008) requieren cumplir 2 o más de los siguientes 7 ítems para un diagnóstico definitivo.
Seis o más manchas café con leche (prepuberales: ≥5 mm de diámetro; pospuberales: ≥15 mm)
Los criterios clínicos incluyen schwannomas vestibulares bilaterales como hallazgo característico. El diagnóstico puede ser más difícil que en NF1 debido a la escasez de síntomas cutáneos. Las pruebas genéticas detectan mutaciones en más del 90% de los casos. La CT/RM confirma neuromas acústicos bilaterales.
Diagnóstico del complejo de esclerosis tuberosa (TSC)
El diagnóstico definitivo se establece con 2 criterios mayores, o 1 criterio mayor y 2 o más criterios menores. La identificación de una variante patogénica de TSC1/TSC2 se reconoce como un criterio diagnóstico independiente 1).
Para evaluar el hamartoma astrocítico retiniano se utilizan el examen de fondo de ojo, la angiografía fluoresceínica y la OCT. El diagnóstico diferencial más importante es el retinoblastoma. La diferenciación es posible por la ausencia de calcificación en la infancia, vasos nutricios escasos y síntomas sistémicos asociados, pero la diferenciación del tumor de mora puede ser difícil.
Según la escala diagnóstica de Roach, se requieren al menos dos de los siguientes: mancha facial en vino de Oporto, elevación de la presión intraocular y angioma leptomeníngeo 1). La OCT de profundidad mejorada y la RM son útiles para evaluar el hemangioma coroideo. La TC de cráneo muestra calcificaciones en la corteza cerebral.
El monitoreo del glaucoma incluye medición regular de la presión intraocular, evaluación del nervio óptico y pruebas de campo visual. En lactantes, además del examen con lámpara de hendidura, pueden ser necesarios el examen de fondo de ojo y la medición de la presión intraocular bajo anestesia.
Las pruebas genéticas pueden detectar mutaciones de VHL en casi el 100% de los casos y son actualmente el método principal para el diagnóstico definitivo. Se recomienda un examen de fondo de ojo con dilatación anual a partir de 1 año de edad. Los hallazgos característicos incluyen tumores esféricos de color naranja rojizo en la retina periférica con vasos dilatados y tortuosos. La angiografía fluoresceínica es útil para detectar hemangioblastomas periféricos pequeños, y la OCT se utiliza para evaluar lesiones pequeñas.
Para el diagnóstico clínico, la tríada de ataxia, telangiectasia conjuntival bulbar y movimientos oculares anormales es importante. Los hallazgos de laboratorio incluyen elevación de AFP sérica, elevación de CA125 y deficiencia de proteína ATM (Western blot). La telangiectasia conjuntival bulbar es un hallazgo patognomónico de esta enfermedad. La RM muestra atrofia cerebelosa difusa en la fosa posterior, particularmente del vermis y los hemisferios 1).
Nódulos de Lisch: Hamartomas asintomáticos que no requieren tratamiento.
Glioma óptico: En casos progresivos se considera la resección quirúrgica, pero se pierde la función visual y hay muchas complicaciones postoperatorias. En casos con infiltración quiasmática está indicada la quimioterapia.
Neurofibroma plexiforme: La extirpación quirúrgica total es difícil y tiende a recurrir. En casos progresivos puede ser necesaria la exenteración orbitaria. En casos no resecables, se usa selumetinib (inhibidor de MEK) como nuevo tratamiento 1).
Glaucoma: El manejo sigue el tratamiento estándar del glaucoma.
Para el schwannoma vestibular, la resección quirúrgica es el estándar; en tumores menores de 3 cm, es posible preservar la audición en el 65% de los casos 1). La queratitis por exposición se maneja con lágrimas artificiales, gafas protectoras corneales y, si es necesario, tarsorrafia.
Tratamiento ocular para el complejo de esclerosis tuberosa (TSC)
Epilepsia: La vigabatrina es el tratamiento de primera línea para las convulsiones parciales infantiles y los espasmos infantiles1). El SEGA (astrocitoma subependimario de células gigantes) es una indicación para resección neuroquirúrgica1).
El tratamiento del glaucoma difiere según la edad de inicio.
Glaucoma congénito (tipo de inicio temprano): El tratamiento quirúrgico es esencial. Se seleccionan trabeculotomía y goniotomía. Se requiere atención cuidadosa al desprendimiento coroideo y la hemorragia, y el riesgo de complicaciones es mayor de lo habitual debido a la elevación de la presión venosa epiescleral. Hay tendencia a la resistencia al tratamiento médico.
Tipo de inicio tardío: primero se realiza tratamiento farmacológico; si es ineficaz, se considera cirugía.
El tratamiento del hemangioma coroideo se selecciona según la presencia o ausencia de cambios exudativos de la siguiente manera.
Fotocoagulación (primera opción): Usando un láser de argón o de colorante, coagule densamente la retina alrededor del hemangioma y los vasos nutricios, luego coagule directamente el tumor. Para tumores de más de 2 diámetros de disco, coagule la arteria aferente y la retina circundante antes de coagular directamente el tumor.
Criocoagulación: Se realiza por vía transescleral cuando la fotocoagulación es difícil debido a la localización periférica del fondo de ojo o a un hemangioma gigante.
Vitrectomía: Se considera cuando los cambios exudativos son graves, se produce desprendimiento de retina o aparecen cambios proliferativos.
Si hay antecedentes familiares, se debe realizar un examen de fondo de ojo a todos los miembros de la familia; el tratamiento temprano puede ofrecer un buen pronóstico.
No existe un tratamiento específico para la telangiectasia conjuntival. Para la inmunodeficiencia, se administran antibióticos profilácticos e inmunoglobulina intravenosa (IVIg) 1). La ataxia se maneja con tratamiento sintomático.
Q¿A qué edad se debe comenzar el cribado de hemangioblastoma retiniano en la VHL?
A
Para VHL, se recomienda un examen de fondo de ojo con dilatación anual a partir de 1 año de edad. Es importante que todos los familiares de pacientes con mutaciones confirmadas del gen VHL también se sometan a un examen de fondo de ojo, ya que el tratamiento temprano puede conducir a un buen pronóstico visual.
Los mecanismos moleculares de cada enfermedad se describen a continuación.
NF1 y TSC (vía mTOR)
NF1 (neurofibromina): Funciona como una RAS-GAP. Promueve la conversión de RAS unido a GTP (activo) a RAS unido a GDP (inactivo). La mutación conduce a la activación persistente de RAS, desregulación de la vía MAP quinasa y la vía PI3K-Akt-mTOR, resultando en proliferación celular descontrolada2).
TSC (hamartina/tuberina): Un complejo supresor de tumores que inhibe directamente los complejos mTOR 1 y 2. La mutación conduce a la sobreactivación de mTOR, causando anomalías en el metabolismo energético, la síntesis de proteínas/lípidos y la supervivencia celular2). Las mutaciones de TSC2 resultan en un fenotipo más grave que las mutaciones de TSC11).
VHL (vía HIF)
pVHL: Componente del complejo E3 ubiquitina ligasa (elongina B/C, Cullin 2, RBX1). En condiciones normales de oxígeno, la prolil hidroxilasa hidroxila HIF-α → pVHL lo reconoce → ubiquitinación → degradación por proteasoma.
Mutación de VHL: Se produce un estado pseudohipóxico constante → acumulación de HIF-α → formación de heterodímero con HIF-1β → activación transcripcional de VEGF, eritropoyesis, metabolismo y genes relacionados con la proliferación celular → formación de tumores vasculares2).
NF2 (merlina): Proteína supresora de tumores expresada en células de Schwann y células meníngeas. Las mutaciones provocan schwannomas, meningiomas y ependimomas.
SWS (mutación mosaico somática de GNAQ): Anomalía en la señalización transmembrana asociada a proteína G. El momento y la ubicación de la mutación en el embrión dan lugar a diferentes patrones de expresión en la región del nervio trigémino, el área intracraneal y el ojo1). GNAQ también es una base molecular común para SWS, KTS y PPV3).
AT (quinasa ATM): Factor supresor de tumores implicado en la reparación de roturas de doble cadena de ADN y el mantenimiento de la estabilidad genómica. Mutación → defecto en la reparación del ADN → predisposición al cáncer, aumento marcado de la radiosensibilidad e inmunodeficiencia1).
Chevalier et al. (2021) examinaron la asociación entre las facomatosis y los tumores endocrinos, e informaron que las vías de señalización de NF1, TSC y VHL (vía RAS-PI3K-Akt-mTOR, vía HIF) están implicadas comúnmente en el desarrollo de neoplasias endocrinas múltiples2).
Q¿Por qué las facomatosis causan lesiones en múltiples órganos?
A
La base fisiopatológica común de las facomatosis es una anomalía en la formación, migración y diferenciación de las células de la cresta neural. Las células de la cresta neural derivan del ectodermo y producen diversas células como células de Schwann y melanocitos, por lo que las lesiones afectan múltiples órganos como el sistema nervioso, la piel y los ojos. Además, las mutaciones en vías de señalización comunes como la vía RAS-mTOR y la vía VHL-HIF promueven el desarrollo de tumores en diferentes órganos.
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
Un fármaco de terapia molecular aprobado por la FDA para neurofibromas plexiformes no resecables. Se dirige a la hiperactivación de la vía MAPK en NF11).
Se están realizando ensayos clínicos para schwannomas relacionados con NF2 y tumores progresivos en el consorcio INTUITT-NF2, con resultados prometedores reportados 1).
Inhibidores de HIF-2α (tumores relacionados con VHL)
Se está llevando a cabo un ensayo de fase 2 dirigido al carcinoma de células renales de células claras asociado a VHL y a los tumores neuroendocrinos pancreáticos.
En el ensayo MK6482 de Chevalier et al. (2021), se observó una respuesta objetiva en el 64% de los tumores neuroendocrinos pancreáticos, con una tasa de supervivencia libre de progresión a 12 meses del 98,3% 2).
Inhibidor de mTOR everolimus (tumores asociados a TSC)
El ensayo EXIST para angiomiolipomas renales y SEGA asociados a TSC confirmó respuestas morfológicas, y también se espera su aplicación en tumores endocrinos 2).
Se ha descubierto que las mutaciones del gen GNAQ son una base molecular común para SWS, el síndrome de Klippel-Trenaunay (KTS) y la facomatosis pigmentovascular (PPV) 3). Debido al riesgo de melanoma coroideo en estos casos superpuestos, se recomiendan exámenes de fondo de ojo con dilatación pupilar 1-2 veces al año 3).
Gama SM, Tamanini JVG, Moraes MPM, Silva TYT, Lima FT, Pedroso JL, et al. A diagnostic approach to neurocutaneous syndromes. Arquivos de neuro-psiquiatria. 2025;83(7):1-14. doi:10.1055/s-0045-1809664. PMID:40562378; PMCID:PMC12196566.
Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Frontiers in endocrinology. 2021;12:678869. doi:10.3389/fendo.2021.678869. PMID:34025587; PMCID:PMC8134657.
Senthilkumar VA, Kohli P, Mishra C, et al. Ocular features in a patient presenting with a rare combination of multiple phakomatoses. BMJ Case Rep. 2022;15:e252746. doi:10.1136/bcr-2022-252746.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.