سندرم نونان (Noonan syndrome; NS) یک بیماری ارثی (RASopathy) است که در اثر جهش در ژنهای مسیر سیگنالدهی RAS-MAPK ایجاد میشود. این سندرم اولین بار در سال ۱۹۶۸ توسط ژاکلین نونان به طور سیستماتیک گزارش شد.
شیوع تخمینی ۱ در ۱۰۰۰ تا ۲۵۰۰ تولد زنده است. الگوی وراثت معمولاً اتوزومال غالب (AD) است، اما حدود دو سوم موارد ناشی از جهشهای جدید (de novo) هستند. جهشهای LZTR1 که به دو شکل AD و AR (اتوزومال مغلوب) دیده میشوند، شناخته شدهاند. در یک خانواده چینی گزارش شده توسط Zhao و همکاران، جهش LZTR1 c.1149+1G>T الگوی وراثت AD را نشان داد1).
«بیان متغیر» که در آن فنوتیپ در یک خانواده با جهش یکسان بسیار متفاوت است و «نفوذ ناقص» که در آن ناقلان فنوتیپ را نشان نمیدهند، مشخصه این بیماری هستند. Han & Park (2024) یک مورد وراثت پدری با جهش PTPN11 p.Arg498Trp گزارش کردند که در آن پدر بدون علامت یا فقط دارای ناتوانی ذهنی خفیف بود و 30 تا 40 درصد خویشاوندان مبتلا بودند2). جهشهای de novo با سن بالای پدر مرتبط هستند و تصور میشود جهشهایی که در طول اسپرماتوژنز رخ میدهند، مزیت انتخابی دارند.
معمولاً به صورت اتوزومال غالب به ارث میرسد، اما حدود دو سوم موارد ناشی از جهشهای de novo (خودبهخودی) است. احتمال انتقال ژن از والد مبتلا به فرزند از نظر تئوری ۵۰٪ است، اما به دلیل نفوذ ناقص، میزان بروز واقعی علائم ۳۰ تا ۴۰٪ تخمین زده میشود2). در صورت وجود سابقه خانوادگی، مشاوره ژنتیک توصیه میگردد.
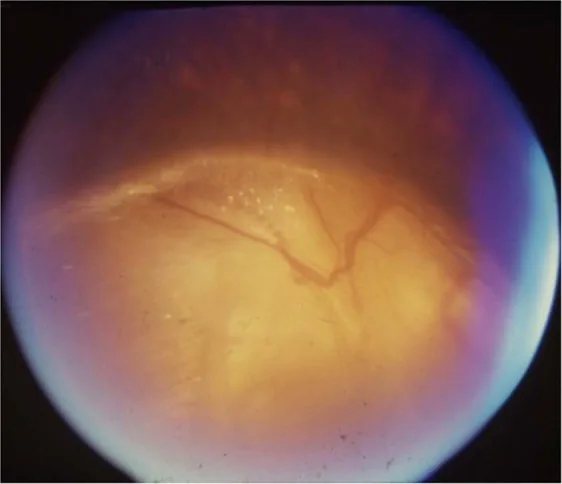
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
چهره مشخصه: پیشانی پهن، فرورفتگی ریشه بینی، گوشهای پایینقرارگرفته و چرخیده به عقب با لاله گوش ضخیم. این ویژگیها در دوران نوزادی و کودکی بارزتر است.
سیستم قلبی عروقی: تنگی دریچه ریوی (۵۰-۶۰٪)، کاردیومیوپاتی هیپرتروفیک (۲۰٪)، نقص دیواره بین دهلیزی (۶-۱۰٪)، نقص دیواره بین بطنی، ناهنجاری لنفاوی.
اسکلت و گردن: گردن بالدار، سینه کبوتری یا قیفی، فاصله زیاد بین نوک پستانها، بزرگی نسبی سر.
خونشناسی: افزایش PT/PTT، اختلال در تعداد و عملکرد پلاکتها، کمبود فاکتور XI (50%). ترومبوسیتوپنی مقاوم در دوره نوزادی میتواند اولین علامت باشد5).
اندام تناسلی: بیضه نزول نکرده در مردان (تا ۸۰٪)، ناباروری.
در مورد Zhao و همکاران (دختر ۶.۶ ساله)، استرابیسم، عیوب انکساری و نیستاگموس مشاهده شد و با کمبود هورمون رشد همراه بود 1). Tian و همکاران (۲۰۲۵) در NS با جهش LZTR1، تغییرات کاورنوماتوز ورید باب، لنفانژکتازی روده و انتروپاتی نشتدهنده پروتئین را گزارش کردند که نشاندهنده تظاهرات متنوع ناهنجاری لنفاوی است 6).
علت NS جهشهای فعالکننده (یا غیرفعالکننده) در چندین ژن دخیل در مسیر RAS-MAPK است. فراوانی ژنهای عامل در زیر نشان داده شده است.
ژن
فراوانی
ویژگیهای اصلی
PTPN11
حدود ۵۰٪
شایعترین. عمدتاً جهشهای افزایش عملکرد
SOS1
حدود ۱۰ تا ۱۳٪
—
RAF1، RIT1
هر کدام حدود ۵٪
ارتباط با اختلال بینایی گزارش شده است
LZTR1
حدود ۸٪
الگوهای توارث اتوزومال غالب و مغلوب
KRAS، BRAF
کمتر از ۵٪ هر کدام
گزارش بدترین حدت بینایی در جهش BRAF
LZTR1 یک پروتئین دستگاه گلژی متعلق به ابرخانواده BTB-Kelch است که با تسریع یوبیکوئیتینسازی و تجزیه RAS، سیگنال RAS-MAPK را به طور منفی تنظیم میکند 3). جهشهای AD-NS در دامنه Kelch (سطح تشخیص بستر) متمرکز شده و سیگنال RAS-MAPK را افزایش میدهند 1)3).
در مورد همبستگی بین ژنوتیپ و فنوتیپ چشمی، گزارشهایی از اختلال بینایی دائمی (بهترین دید اصلاحشده دوچشمی <0.3) در بیماران با جهشهای RAF1، KRAS و SHOC2، و بدترین دید (بینایی ضعیف از دوران نوزادی به دلیل هیپوپلازی عصب بینایی) در بیماران با جهش BRAF وجود دارد، اما همبستگی قطعی هنوز مشخص نشده و گروههای مطالعه کوچک (25 تا 105=n) هستند.
در NFNS (سندرم نوروفیبروماتوز-نونان) ناشی از جهش NF1، حدود 25٪ از بیماران NF1 ویژگیهای شبه نونان را نشان میدهند که فنوتیپ دو بیماری همپوشانی دارد 4).
Qآیا شدت علائم چشمی بسته به نوع ژن متفاوت است؟
A
در بیماران با جهشهای RAF1، KRAS و SHOC2، نابینایی دائمی گزارش شده است و در بیماران با جهش BRAF، ضعف بینایی از دوران نوزادی به دلیل هیپوپلازی عصب بینایی گزارش شده است. از سویدیگر، برخی گزارشها نشان میدهد که در بیماران با جهش PTPN11 اختلال بینایی مشاهده نشده است. با این حال، این همبستگیها در گروههای کوچک مشاهده شده و در حال حاضر همبستگی قطعی ژنوتیپ-فنوتیپ ثابت نشده است.
تشخیص NS بر اساس معیارهای رسمی تشخیصی مبتنی بر یافتههای بالینی مانند چهره مشخص، بیماری قلبی و کوتاهی قد، توسط متخصص ژنتیک بالینی ارزیابی میشود.
برای تشخیص قطعی از آزمایش ژنتیکی مولکولی با استفاده از پانل ژنهای مرتبط با مسیر RAS-MAPK (شامل PTPN11، SOS1، RAF1، RIT1، KRAS، LZTR1 و غیره) استفاده میشود2)3). همچنین توالییابی کامل اگزوم (WES) به کار میرود1)4). توجه داشته باشید که ممکن است LZTR1 در پانل ژنی NS گنجانده نشده باشد3).
نسبت DM/DD (فاصله ماکولا-دیسک به قطر دیسک): نسبت ۳ یا بیشتر نشاندهنده مشکوک بودن به هیپوپلازی عصب بینایی است و نسبت ۴ یا بیشتر احتمال آن را افزایش میدهد.
NFNS (سندرم نوروفیبروماتوز-نونان): جهش NF1 باعث همپوشانی فنوتیپ NS+NF1 میشود. در صورت وجود ۶ یا بیشتر لکههای کافهاو له یا اندازه ۵ میلیمتر یا بیشتر، به دنبال همراهی NF1 باشید4). Farncombe و همکاران نوروفیبروم پلکسیفرم را در بیماران NS با جهش LZTR1 مشاهده کرده و همپوشانی بالینی با NF1 را گزارش کردند3).
تشخیص افتراقی کولوبومای سر عصب بینایی: استافیلوم اطراف پاپی، سندرم گل صبحگاهی، PFV/PHPV پاپیلاری، ماکروپاپی.
اصلاح عیوب انکساری: اصلاح عیوب انکساری با استفاده از عینک یا لنزهای تماسی.
درمان تنبلی چشم: درمان تنبلی چشم با استفاده از پوشاندن چشم سالم و روشهای مشابه.
جراحی استرابیسم: بسته به میزان انحراف چشم، جراحی در نظر گرفته میشود.
هیپوپلازی عصب بینایی: غیرپیشرونده است و در صورت عدم همراهی با گلوکوم، دید و میدان بینایی تغییر نمیکند. از کاهش فشار چشم بهطور خودسرانه خودداری کنید. با اصلاح مناسب عیوب انکساری، سعی در بهبود عملکرد بینایی باقیمانده کنید.
جداشدگی سروزی شبکیه همراه با کولوبومای سر عصب بینایی: درمان مشخصی وجود ندارد و در برخی موارد خودبهخود بهبود مییابد. در جداشدگی رگماتوژن شبکیه، درمان جراحی مشابه آن در نظر گرفته میشود.
کوتاهی قد: درمان با هورمون رشد (GH). در برخی بیماران NS کمبود GH همراه است 1). با این حال، در NFNS1 تجویز GH خطر افزایش نوروفیبروم را دارد و نیاز به تصمیمگیری محتاطانه دارد 4).
بیماریهای قلبی: بالنگشایی یا ترمیم جراحی برای تنگی دریچه ریوی. پیگیری و درمان دارویی برای کاردیومیوپاتی هیپرتروفیک. کاردیومیوپاتی هیپرتروفیک در کودکان زیر ۲ سال بالاترین خطر مرگ قلبی را دارد5).
ناهنجاری خونی: جایگزینی فاکتورهای انعقادی. ارزیابی خطر خونریزی قبل از عمل. در نوزاد گزارششده توسط Tang و همکاران، ترومبوسیتوپنی مقاوم اولین علامت بود و با نقص دیواره بیندهلیزی و شیلوتوراکس همراه بود5).
شیلوتوراکس (افیوژن شیلوس): درناژ قفسه سینه. در مطالعه Tian و همکاران، جراحی میکروسکوپی برای انسداد مجرای توراسیک منجر به بهبود هیپوآلبومینمی شد6).
بیضه نزولنکرده: جراحی تثبیت بیضه در زمان مناسب.
Qدرمان چشم در سندرم نونان چگونه انجام میشود؟
A
تصحیح عیوب انکساری با عینک یا لنز تماسی و درمان تنبلی چشم اساس درمان است. برای استرابیسم (انحراف چشم) بسته به شدت، جراحی در نظر گرفته میشود. هیپوپلازی عصب بینایی غیرپیشرونده است و از کاهش فشار چشم بدون دلیل باید خودداری کرد؛ با تصحیح مناسب عیوب انکساری، بهبود عملکرد بینایی باقیمانده هدف قرار میگیرد. جداشدگی سروزی شبکیه همراه با کولوبومای دیسک بینایی گاهی خودبهخود بهبود مییابد و درمان مشخصی وجود ندارد.
پاتوفیزیولوژی اصلی NS، انتقال سیگنال پایدار و بیش از حد ناشی از اختلال تنظیم مسیر RAS-MAPK است.
PTPN11 (SHP2)
عملکرد: تیروزین فسفاتاز غیرگیرندهای. دارای مکانیسم خودتنظیمی از طریق برهمکنش درونمولکولی دامنه N-SH2 و دامنه PTP.
تأثیر جهش NS: مکانیسم خودمهارگری غیرفعال شده و فعالیت فسفاتاز افزایش مییابد (کسب عملکرد). سیگنالدهی RAS-MAPK بیش از حد فعال میشود 2).
تفاوت با NSML: جهشهای NSML (سندرم لئوپارد) از نوع کاهشدهنده فعالیت PTPN11 هستند که در تضاد با NS میباشند.
LZTR1
عملکرد: پروتئین دستگاه گلژی متعلق به ابرخانواده BTB-Kelch. پلییوبیکوئیتینه کردن و تجزیه پروتئازومی RAS را تسریع کرده و سیگنال RAS-MAPK را به صورت منفی تنظیم میکند3).
جهشهای AD-NS: متمرکز در دامنه Kelch (سطح تشخیص سوبسترا). باعث افزایش سیگنالدهی RAS-MAPK (نوع عملکردی افزایشیافته) میشوند 1)3).
جهش AR-NS: ناشی از جهشهای از دستدهنده عملکرد. همچنین دارای عملکرد اتصال به کمپلکس RAF1/SHOC2/PP1CB و تسریع فسفریلاسیون Ser259 RAF1 (غیرفعالسازی سیگنال MAPK) است3).
اجزای اصلی مسیر RAS-MAPK به شرح زیر است.
تنظیمکنندههای مثبت: SHP2 (PTPN11)، SOS1، SOS2. جهشهای افزایش عملکردی این ژنها باعث ایجاد NS میشوند.
عوامل تقویتکننده سیگنال RAS: MRAS، SHOC2، PPP1CB.
آبشار MAPK: BRAF، RAF1، MAP2K1، MAP2K2، MAPK1.
تنظیمکنندههای منفی: CBL، NF1، LZTR1، SPRED1، SPRED2. جهشهای از دست دادن عملکرد باعث افزایش سیگنالدهی میشوند3).
مسیر RAS-MAPK برای تمایز و تکثیر سلولی بسیار حیاتی است و جهشهای افزایش عملکرد باعث ایجاد عملکرد غیرطبیعی سلولی در بافتهای سراسر بدن شده و منجر به فنوتیپهای پیچیده در چندین اندام میشود.
از نظر پاتوفیزیولوژی چشمی، هیپوپلازی عصب بینایی ناشی از عدم تکامل سلولهای گانگلیونی شبکیه و رشتههای عصبی است. کولوبومای سر عصب بینایی ناشی از بسته نشدن شکاف کاسه چشم است و شیوع آن ۳ تا ۸ در ۱۰۰٬۰۰۰ تخمین زده میشود.
در ارتباط با تومورها، جهش PTPN11 به عنوان جهش سوماتیک در JMML (لوسمی میلومونوسیتی جوانان)، MDS و AML نیز شناسایی شده است و NS خطر بدخیمیهای خونی دوران کودکی را سه برابر افزایش میدهد 5).
7. تحقیقات جدید و چشماندازهای آینده (گزارشهای مرحله تحقیقاتی)
Tian و همکاران (2025) در یک مورد سندرم نونان با جهش LZTR1 c.850C>T، تغییرات کاورنوماتوز ورید باب، ناهنجاری مجرای توراسیک، اتساع عروق لنفاوی روده و انتروپاتی نشتدهنده پروتئین را گزارش کردند6). جراحی میکروسکوپی برای انسداد خروجی مجرای توراسیک انجام شد و نرمالسازی آلبومین سرم حاصل گردید. این یافته نشاندهنده پاتوفیزیولوژی جدیدی از ناهنجاری لنفاوی همراه با سندرم نونان ناشی از جهش LZTR1 است.
Orsolini و همکاران (2024) در یک مرد 35 ساله با جهش LZTR1 c.742G>A، بدون سابقه بیضه نزولنکرده، FSH بالا و الیگواسپرمی (غلظت اسپرم 1.5×10⁶/mL) را گزارش کردند7). این مورد وجود اختلال عملکرد گناد ناشی از آسیب اولیه سلولهای سرتولی را مستقل از بیضه نزولنکرده نشان میدهد و یافتهای است که به روشنسازی مکانیسم ناباروری مردانه در سندرم نونان کمک میکند.
LZTR1 همچنین ژن عامل شوانوماتوز (schwannomatosis) است و تحقیقات در مورد همپوشانی بالینی NS، NF1 و شوانوماتوز در حال پیشرفت است3). رابطه بیولوژیکی بین RIT1 و LZTR1 نیز مورد توجه قرار گرفته است و احتمال داده میشود که جهش در هر یک از این دو باعث تجمع RIT1 و کمک به بیشفعالی مسیر MAPK شود3).
در مورد همبستگی ژنوتیپ-فنوتیپ، ایجاد آن با استفاده از گروههای بزرگتر یک چالش آینده است. شناسایی عوامل پیشبینیکننده پیشآگهی چشمی و توسعه اهداف درمانی اختصاصی برای سندرم نونان مورد انتظار است.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.