Noonan syndrome (NS) is a genetic disorder (RASopathy) caused by gene mutations in the RAS-MAPK signaling pathway. It was first systematically reported by Jacqueline Noonan in 1968.
The estimated prevalence is 1 in 1,000 to 2,500 births. The inheritance pattern is usually autosomal dominant (AD), but about two-thirds of cases are due to de novo mutations. LZTR1 mutations can be both AD and autosomal recessive (AR); in a Chinese family reported by Zhao et al., the LZTR1 c.1149+1G>T mutation showed AD inheritance 1).
Variable expressivity, where the phenotype differs greatly within families carrying the same mutation, and incomplete penetrance, where carriers do not show the phenotype, are characteristic. Han & Park (2024) reported a paternal inheritance case with the PTPN11 p.Arg498Trp mutation; the father was asymptomatic or had only mild intellectual disability, and the penetrance among affected relatives is estimated at 30–40% 2). De novo mutations are associated with advanced paternal age, and it is thought that mutations arising during spermatogenesis have a selective advantage.
It usually shows autosomal dominant inheritance, but about two-thirds of cases are due to de novo mutations. The theoretical risk of transmission from an affected parent to a child is 50%, but due to incomplete penetrance, the actual rate of symptoms is estimated at 30–40% 2). Genetic counseling is recommended if there is a family history.
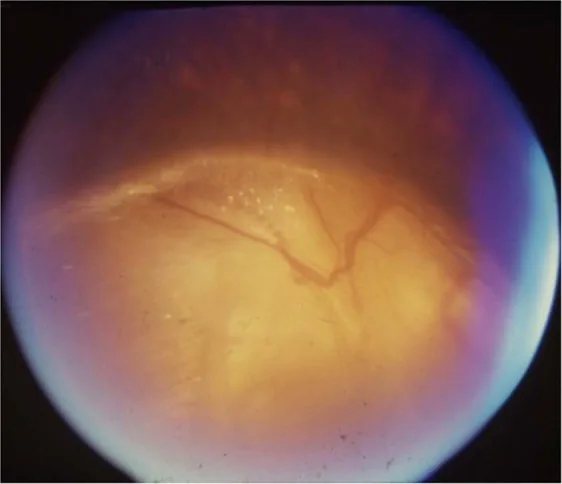
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Noonan syndrome is characterized by distinctive facial features, short stature, and congenital heart defects.
Hematologic: Prolonged PT/PTT, abnormal platelet count and function, factor XI deficiency (50%). Refractory thrombocytopenia in the neonatal period may be the initial presentation5).
Genitalia: Cryptorchidism in males (up to 80%), infertility.
Zhao et al. reported a case (6.6-year-old girl) with strabismus, refractive error, and nystagmus, complicated by growth hormone deficiency 1). Tian et al. (2025) reported portal cavernous transformation, intestinal lymphangiectasia, and protein-losing enteropathy in LZTR1-mutated NS, demonstrating diverse lymphatic malformations 6).
The cause of NS is gain-of-function (or loss-of-function) mutations in multiple genes involved in the RAS-MAPK pathway. The frequencies of causative genes are shown below.
Gene
Frequency
Main features
PTPN11
Approximately 50%
Most common. Mainly gain-of-function mutations.
SOS1
Approximately 10–13%
—
RAF1, RIT1
Approximately 5% each
Reported association with visual impairment
LZTR1
Approximately 8%
Both AD and AR inheritance patterns
KRAS, BRAF
Less than 5% each
BRAF mutation associated with reports of worst visual acuity
LZTR1 is a Golgi protein belonging to the BTB-Kelch superfamily that promotes ubiquitination and degradation of RAS, thereby negatively regulating RAS-MAPK signaling 3). Mutations in AD-NS are concentrated in the Kelch domain (substrate recognition surface) and enhance RAS-MAPK signaling 1)3).
Regarding genotype-ophthalmic phenotype correlations, reports indicate permanent visual impairment (best-corrected visual acuity < 0.3 in both eyes) in patients with RAF1, KRAS, and SHOC2 mutations, and the worst visual acuity (poor vision from infancy due to optic nerve hypoplasia) in patients with BRAF mutations. However, definitive correlations have not been established, and cohorts are small (n=25–105).
In NFNS (neurofibromatosis-Noonan syndrome) caused by NF1 mutations, approximately 25% of NF1 patients exhibit Noonan-like features, with overlapping phenotypes of both conditions 4).
QDo eye symptoms vary depending on the type of gene?
A
Permanent visual impairment has been reported in patients with RAF1, KRAS, and SHOC2 mutations, and poor vision from infancy due to optic nerve hypoplasia has been reported in patients with BRAF mutations. On the other hand, some reports indicate that visual impairment was not observed in patients with PTPN11 mutations. However, the cohorts showing these correlations are small, and a definitive genotype-phenotype correlation has not been established at present.
The diagnosis of NS is evaluated by a clinical geneticist according to formal diagnostic criteria based on clinical findings such as characteristic facial features, heart disease, and short stature.
For definitive diagnosis, molecular genetic testing using a RAS-MAPK pathway-related gene panel (including PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1, etc.) is used2)3). Whole exome sequencing (WES) is also used1)4). Note that LZTR1 may not be included in some NS gene panels3).
NFNS (Neurofibromatosis-Noonan syndrome): NF1 mutation causes overlapping phenotypes of NS and NF1. Search for NF1 comorbidity if there are 6 or more café-au-lait spots or spots ≥5 mm in size 4). Farncombe et al. reported plexiform neurofibromas in NS patients with LZTR1 mutation, indicating clinical overlap with NF13).
Refractive correction: Correction of refractive errors with glasses or contact lenses.
Amblyopia treatment: Treatment of amblyopia using occlusion of the healthy eye, etc.
Strabismus surgery: Surgery is considered depending on the degree of strabismus.
Optic nerve hypoplasia: This is non-progressive, and vision and visual fields do not change unless glaucoma is present. Avoid unnecessary intraocular pressure-lowering treatment. Attempt to improve residual visual function with appropriate refractive correction.
Serous retinal detachment associated with optic disc coloboma: There is no established treatment, and spontaneous resolution may occur. For rhegmatogenous retinal detachment, consider surgical treatment accordingly.
Short stature: Growth hormone (GH) therapy. Some NS patients have GH deficiency 1). However, in NFNS1, GH administration carries a risk of neurofibroma enlargement, requiring careful consideration 4).
Heart disease: Balloon dilation or surgical repair for pulmonary valve stenosis. Observation and medication for hypertrophic cardiomyopathy. Hypertrophic cardiomyopathy in children under 2 years of age carries the highest risk of cardiac death 5).
Blood abnormalities: Coagulation factor replacement. Preoperative bleeding risk assessment. In the neonatal case by Tang et al., refractory thrombocytopenia was the initial symptom, and the patient had concurrent ASD and chylothorax 5).
Chylothorax: Chest tube drainage. In Tian et al., microsurgery for thoracic duct outlet obstruction improved hypoalbuminemia 6).
Undescended testis: Orchiopexy at the appropriate time.
QHow is eye treatment performed for Noonan syndrome?
A
The basics are refractive correction with glasses or contact lenses and amblyopia treatment. Surgery for strabismus is considered depending on the degree. Optic nerve hypoplasia is non-progressive; avoid unnecessary intraocular pressure-lowering treatment and aim to improve residual visual function with appropriate refractive correction. Serous retinal detachment associated with optic disc coloboma may resolve spontaneously, and there is no established treatment.
The fundamental pathophysiology of NS is sustained and excessive signal transduction due to dysregulation of the RAS-MAPK pathway.
PTPN11 (SHP2)
Function: Non-receptor type tyrosine phosphatase. It has an autoinhibitory mechanism through intramolecular interaction between the N-SH2 domain and the PTP domain.
Effect of NS mutations: The autoinhibitory mechanism is released, leading to increased phosphatase activity (gain-of-function). RAS-MAPK signaling becomes hyperactivated 2).
Difference from NSML: NSML (LEOPARD syndrome) mutations are loss-of-function in PTPN11, in contrast to NS.
LZTR1
Function: A Golgi protein belonging to the BTB-Kelch superfamily. It promotes polyubiquitination and proteasomal degradation of RAS, negatively regulating RAS-MAPK signaling 3).
AD-NS mutations: Concentrated in the Kelch domain (substrate recognition surface). They enhance RAS-MAPK signaling (gain-of-function) 1)3).
AR-NS mutation: Loss-of-function mutation. It also binds to the RAF1/SHOC2/PP1CB complex and promotes RAF1 Ser259 phosphorylation (inactivating MAPK signaling)3).
The main components of the RAS-MAPK pathway are as follows.
Positive regulators: SHP2 (PTPN11), SOS1, SOS2. Gain-of-function mutations in these cause NS.
RAS signaling promoters: MRAS, SHOC2, PPP1CB.
MAPK cascade: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Negative regulators: CBL, NF1, LZTR1, SPRED1, SPRED2. Loss-of-function mutations cause signal enhancement3).
The RAS-MAPK pathway is crucial for cell differentiation and proliferation. Gain-of-function mutations cause abnormal cellular function in systemic tissues, leading to complex phenotypes involving multiple organs.
Ophthalmologically, optic nerve hypoplasia results from developmental failure of retinal ganglion cells and nerve fibers. Optic disc coloboma is caused by incomplete closure of the optic fissure, with a prevalence of 3–8 per 100,000.
Regarding tumors, PTPN11 mutations are also identified as somatic mutations in JMML (juvenile myelomonocytic leukemia), MDS, and AML. NS is associated with a threefold increased risk of childhood hematologic malignancies 5).
7. Latest Research and Future Perspectives (Research-stage Reports)
Tian et al. (2025) reported a case of LZTR1 c.850C>T mutant NS with portal cavernous transformation, thoracic duct dysplasia, intestinal lymphangiectasia, and protein-losing enteropathy6). Microsurgery for thoracic duct outlet obstruction was performed, achieving normalization of serum albumin. This finding reveals a new pathology of lymphatic malformations associated with LZTR1-mutant NS.
Orsolini et al. (2024) reported a 35-year-old male with LZTR1 c.742G>A mutation who had elevated FSH and oligospermia (sperm concentration 1.5×10⁶/mL) without a history of cryptorchidism7). This case suggests the presence of gonadal dysfunction due to primary Sertoli cell injury independent of cryptorchidism, contributing to the elucidation of the mechanism of male infertility in NS.
Genotype-phenotype correlation and future challenges
LZTR1 is also a causative gene for schwannomatosis, and research on the clinical overlap among NS, NF1, and schwannomatosis is progressing 3). The biological relationship between RIT1 and LZTR1 is also attracting attention, and it has been suggested that mutations in either may cause RIT1 accumulation and contribute to overactivation of the MAPK signaling pathway 3).
Regarding genotype-phenotype correlations, establishing them through larger cohorts remains a future challenge. Identification of predictors for ophthalmic prognosis and development of NS-specific therapeutic targets are expected.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.