眼科所見

努南症候群
一目瞭然的要點
Section titled “一目瞭然的要點”1. 什麼是努南症候群?
Section titled “1. 什麼是努南症候群?”努南症候群(Noonan syndrome; NS)是一種由RAS-MAPK訊息傳導路徑基因突變引起的遺傳性疾病(RASopathy)。1968年由Jacqueline Noonan首次系統性報告。
估計盛行率為每1,000至2,500名新生兒中1例。遺傳方式通常為體染色體顯性遺傳(AD),但約三分之二的病例是由新發突變引起。已知LZTR1突變可表現為AD和體染色體隱性遺傳(AR)兩種形式;在Zhao等人報導的中國家系中,LZTR1 c.1149+1G>T突變呈AD遺傳1)。
同一突變的家系內表現型差異很大,即「可變表現度」,以及帶因者不表現表現型的「不完全外顯率」是其特點。Han & Park(2024)報導了一例父系遺傳的PTPN11 p.Arg498Trp突變病例;父親無症狀或僅有輕度智能障礙,受累親屬的外顯率估計為30–40%2)。新發突變與父親高齡相關,認為精子形成過程中產生的突變具有選擇性優勢。
眼科方面,可能伴隨視神經發育不全、視盤缺損、屈光異常、眼瞼下垂等多種異常。
Q
努南氏症的遺傳機率是多少?
A
通常為體染色體顯性遺傳,但約三分之二的病例是由新發突變引起。罹病父母傳給子女的理論風險為50%,但由於不完全外顯率,實際出現症狀的比例估計為30–40%2)。有家族史者建議進行遺傳諮詢。
2. 主要症狀與臨床所見
Section titled “2. 主要症狀與臨床所見”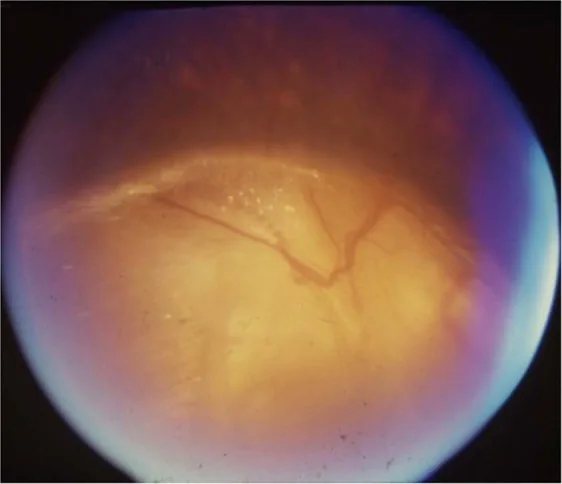
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
努南氏症候群的特徵包括特殊面容、身材矮小和先天性心臟缺陷。
- 視力下降:因視神經發育不全或缺損,嬰兒期可能就有視力不良。
- 斜視:自覺眼位偏移。兒童期常由父母發現。
- 身材矮小:出生時身高正常,但隨著成長,身材矮小變得明顯。
- 發展遲緩:語言發展遲緩和輕度智能障礙可能是初期主訴。
全身表現
Zhao等人的病例(6.6歲女童)出現斜視、屈光異常和眼震,並合併生長激素缺乏1)。Tian等人(2025)報告了LZTR1突變NS患者出現門靜脈海綿狀變性、腸淋巴管擴張和蛋白質流失性腸病,顯示淋巴管發育異常的多樣表現6)。
Q
努南氏症候群的眼部症狀有哪些?
3. 原因與風險因素
Section titled “3. 原因與風險因素”NS的病因是RAS-MAPK路徑中多個基因的功能獲得性(或功能喪失性)突變。以下列出致病基因的頻率。
| 基因 | 頻率 | 主要特徵 |
|---|---|---|
| PTPN11 | 約50% | 最常見。主要為功能獲得型突變。 |
| SOS1 | 約10~13% | — |
| RAF1, RIT1 | 各約5% | 有報告與視力障礙相關 |
| LZTR1 | 約8% | AD和AR兩種遺傳形式 |
| KRAS, BRAF | 各低於5% | BRAF突變有最低視力的報告 |
LZTR1是一種屬於BTB-Kelch超家族的高基氏體蛋白,透過促進RAS的泛素化與降解來負向調控RAS-MAPK訊息傳遞3)。AD-NS的突變集中在Kelch結構域(受質辨識面),進而增強RAS-MAPK訊息傳遞1)3)。
關於基因型與眼科表現型的相關性,有報告指出RAF1、KRAS和SHOC2突變患者出現永久性視力障礙(雙眼最佳矯正視力<0.3),BRAF突變患者視力最差(因視神經發育不全導致嬰兒期視力不良)。但尚未建立確定的相關性,且研究隊列規模較小(n=25~105)。
在由NF1突變引起的NFNS(神經纖維瘤病-努南症候群)中,約25%的NF1患者表現出努南樣特徵,兩種疾病的表現型重疊4)。
Q
眼睛症狀的嚴重程度是否因基因類型而異?
A
在RAF1、KRAS、SHOC2突變患者中已有永久性視力障礙的報告,而在BRAF突變患者中,因視神經發育不全導致嬰兒期視力不良也有報告。另一方面,有報告指出PTPN11突變患者未觀察到視力障礙。然而,顯示這些相關性的隊列規模較小,目前尚未建立確定的基因型-表現型相關性。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”臨床診斷與遺傳學檢查
Section titled “臨床診斷與遺傳學檢查”NS的診斷由臨床遺傳學專家根據基於特徵性面容、心臟疾病、身材矮小等臨床發現的正式診斷標準進行評估。
確診採用RAS-MAPK路徑相關基因面板(包括PTPN11、SOS1、RAF1、RIT1、KRAS、LZTR1等)進行分子遺傳學檢測2)3)。全外顯子組測序(WES)也可使用1)4)。需注意NS基因面板可能不包含LZTR13)。
診斷時建議進行全面的眼科評估,之後每年追蹤一次。
- 屈光檢查:評估屈光不正。檢查是否有高度屈光不正和散光。
- 斜視檢查:評估眼位和眼球運動。
- 眼底檢查:評估視神經盤形態。
視神經發育不全的評估
Section titled “視神經發育不全的評估”- DM/DD比值(視盤黃斑距離/視盤直徑比):≥3時懷疑視神經發育不全,≥4時可能性高。
- 雙環徵:視乳頭周圍色素環。
- 光學同調斷層掃描(OCT):測量cpRNFL(視乳頭周圍視網膜神經纖維層)厚度有幫助。
- 頭部MRI:評估中樞神經系統異常。約15%有腦下垂體漏斗異常。
視神經盤缺損的診斷
Section titled “視神經盤缺損的診斷”- 檢眼鏡所見:以下方為中心的視乳頭及視網膜脈絡膜缺損、血管走向異常。
- 影像檢查:透過超音波、MRI、CT、OCT確診。頭部MRI/CT檢查有無顱內畸形。
- NFNS(神經纖維瘤病-努南症候群):NF1突變導致NS和NF1表型重疊。若有6個以上或直徑≥5mm的咖啡斑,需檢查是否合併NF14)。Farncombe等人報告LZTR1突變NS患者出現叢狀神經纖維瘤,顯示與NF1的臨床重疊3)。
- 視盤缺損的鑑別:視盤周圍葡萄腫、晨光症候群、視盤PFV/PHPV、巨大視盤。
- 其他症候群:CHARGE症候群、透納症候群、心臉皮膚症候群(CFC)、Costello症候群、Legius症候群。
- 心臟超音波:心臟畸形篩檢。
- 血液檢查:凝血功能檢查(PT/APTT、第XI因子活性等)5)。
5. 標準治療方法
Section titled “5. 標準治療方法”NS沒有根治性治療,以臨床遺傳學專家為中心的多學科協作管理是基礎。
- 屈光矯正:透過眼鏡或隱形眼鏡矯正屈光不正。
- 弱視治療:使用遮蓋健眼等方法治療弱視。
- 斜視手術:根據斜視程度考慮手術。
- 視神經發育不全:為非進行性,若不併發青光眼,視力和視野不會改變。避免輕易進行降眼壓治療。透過適當的屈光矯正嘗試改善殘餘視功能。
- 視盤缺損伴發的漿液性視網膜剝離:尚無確定的治療方法,也有自然消退的病例。對於裂孔源性視網膜剝離,考慮相應的手術治療。
- 身材矮小:生長激素(GH)治療。部分NS患者合併GH缺乏1)。但在NFNS1中,GH給藥存在神經纖維瘤增大的風險,需謹慎判斷4)。
- 心臟疾病:肺動脈瓣狹窄的氣球擴張術或外科修復。肥厚型心肌病的觀察和藥物治療。2歲以下肥厚型心肌病患兒的心臟性死亡風險最高5)。
- 血液異常:補充凝血因子。術前出血風險評估。Tang等人的新生兒案例中,頑固性血小板減少為初始症狀,合併ASD和乳糜胸5)。
- 乳糜胸:胸腔引流。Tian等人透過顯微手術治療胸管出口阻塞,改善了低蛋白血症6)。
- 隱睪:適時進行睪丸固定術。
Q
努南症候群的眼部治療如何進行?
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”NS的根本病理生理是RAS-MAPK路徑調節異常導致的持續過度訊號傳導。
PTPN11(SHP2)
功能:非受體型酪胺酸磷酸酶。透過N-SH2結構域和PTP結構域之間的分子內相互作用具有自身抑制機制。
NS突變的影響:自身抑制機制被解除,磷酸酶活性增強(功能獲得型)。RAS-MAPK訊息過度活化2)。
與NSML的差異:NSML(豹皮症候群)突變是PTPN11的活性降低型,與NS相反。
LZTR1
功能:屬於BTB-Kelch超家族的高基氏體蛋白。促進RAS的多聚泛素化和蛋白酶體降解,負調控RAS-MAPK訊息3)。
AD-NS的突變:集中在Kelch結構域(受質辨識面)。增強RAS-MAPK訊息(功能獲得型)1)3)。
AR-NS突變:功能喪失型突變。它還能結合RAF1/SHOC2/PP1CB複合物並促進RAF1 Ser259磷酸化(使MAPK信號失活)3)。
RAS-MAPK路徑的主要組成部分如下。
- 正調控因子:SHP2(PTPN11)、SOS1、SOS2。這些因子的功能獲得型突變導致NS。
- RAS信號促進因子:MRAS、SHOC2、PPP1CB。
- MAPK級聯:BRAF、RAF1、MAP2K1、MAP2K2、MAPK1。
- 負調控因子:CBL、NF1、LZTR1、SPRED1、SPRED2。功能喪失型突變導致信號增強3)。
RAS-MAPK路徑對細胞分化與增生至關重要。功能獲得型突變導致全身組織細胞功能異常,引起涉及多器官的複雜表現型。
眼科方面,視神經發育不全由視網膜神經節細胞與神經纖維發育障礙引起。視盤缺損由眼杯裂閉合不全導致,盛行率為3~8/100,000。
與腫瘤相關,PTPN11突變也被鑑定為JMML(幼年型粒單核細胞白血病)、MDS和AML的體細胞突變。NS患童血液惡性腫瘤風險增加3倍5)。
7. 最新研究與未來展望(研究階段報告)
Section titled “7. 最新研究與未來展望(研究階段報告)”LZTR1突變NS中的淋巴管形成異常
Section titled “LZTR1突變NS中的淋巴管形成異常”Tian等人(2025)報告了一例LZTR1 c.850C>T突變NS病例,伴有門靜脈海綿狀變性、胸導管發育不全、腸淋巴管擴張和蛋白質流失性腸病6)。對胸導管出口阻塞進行了顯微手術,實現了血清白蛋白的正常化。這一發現揭示了LZTR1突變NS相關淋巴管形成異常的新病理機制。
男性不育的機制
Section titled “男性不育的機制”Orsolini等人(2024)報告了一名攜帶LZTR1 c.742G>A突變的35歲男性,無隱睪病史,但出現高FSH和少精子症(精子濃度1.5×10⁶/mL)7)。該病例提示存在獨立於隱睪的原發性支持細胞損傷導致的性腺功能障礙,有助於闡明NS中男性不育的機制。
基因型-表現型相關性及未來挑戰
Section titled “基因型-表現型相關性及未來挑戰”LZTR1也是神經鞘瘤病(schwannomatosis)的致病基因,關於NS、NF1和神經鞘瘤病臨床重疊的研究正在進展中3)。RIT1與LZTR1的生物學關係也備受關注,有研究指出其中任一基因的突變可能導致RIT1累積,進而促進MAPK訊息傳導路徑的過度活化3)。
關於基因型-表現型關聯,透過更大規模的世代研究來確立是未來的課題。預期能識別眼科預後的預測因子,並開發NS特異性的治療標靶。
8. 參考文獻
Section titled “8. 參考文獻”- Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
- Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
- Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
- Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
- Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
- Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
- Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.