El síndrome de Noonan (NS) es un trastorno genético (RASopatía) causado por mutaciones en genes de la vía de señalización RAS-MAPK. Fue descrito sistemáticamente por primera vez por Jacqueline Noonan en 1968.
La prevalencia estimada es de 1 por cada 1,000 a 2,500 nacimientos. El patrón de herencia suele ser autosómico dominante (AD), pero aproximadamente dos tercios de los casos se deben a mutaciones de novo. Se conocen mutaciones en LZTR1 que pueden ser tanto AD como autosómico recesivo (AR); en una familia china reportada por Zhao et al., la mutación LZTR1 c.1149+1G>T mostró herencia AD 1).
Son características la expresividad variable, donde el fenotipo difiere mucho dentro de familias con la misma mutación, y la penetrancia incompleta, donde los portadores no muestran el fenotipo. Han & Park (2024) reportaron un caso de herencia paterna con la mutación PTPN11 p.Arg498Trp; el padre estaba asintomático o solo tenía discapacidad intelectual leve, y la penetrancia entre familiares afectados se estima en 30–40% 2). Las mutaciones de novo se asocian con la edad paterna avanzada, y se cree que las mutaciones que surgen durante la espermatogénesis tienen una ventaja selectiva.
Q¿Con qué frecuencia se hereda el síndrome de Noonan?
A
Generalmente muestra herencia autosómica dominante, pero aproximadamente dos tercios de los casos se deben a mutaciones de novo. El riesgo teórico de transmisión de un padre afectado a un hijo es del 50%, pero debido a la penetrancia incompleta, la tasa real de síntomas se estima en 30–40% 2). Se recomienda asesoramiento genético si hay antecedentes familiares.
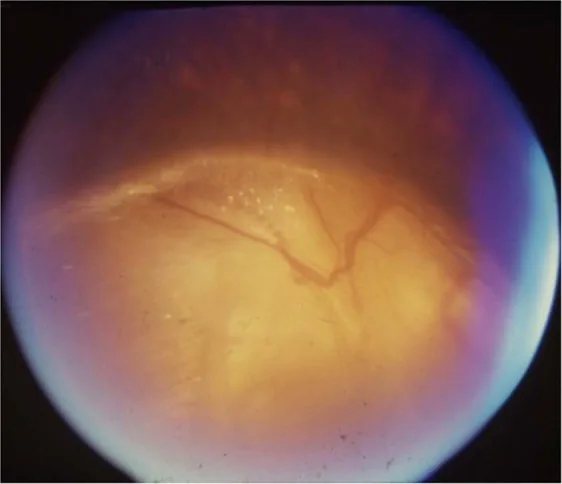
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
El síndrome de Noonan se caracteriza por rasgos faciales distintivos, baja estatura y defectos cardíacos congénitos.
Rasgos faciales característicos: Frente ancha, puente nasal hundido, orejas de implantación baja y rotadas posteriormente con hélix grueso. Más prominente en la infancia y niñez.
Cardiovascular: Estenosis de la válvula pulmonar (50–60%), miocardiopatía hipertrófica (20%), CIA (6–10%), CIV, malformaciones linfáticas.
Hematológico: TP/TTPa prolongados, alteraciones en el recuento y función plaquetaria, deficiencia del factor XI (50%). La trombocitopenia refractaria en el período neonatal puede ser la presentación inicial5).
Genitales: Criptorquidia en varones (hasta el 80%), infertilidad.
Zhao et al. reportaron un caso (niña de 6.6 años) con estrabismo, error refractivo y nistagmo, complicado con deficiencia de hormona de crecimiento 1). Tian et al. (2025) reportaron transformación cavernosa portal, linfangiectasia intestinal y enteropatía perdedora de proteínas en NS con mutación de LZTR1, demostrando diversas manifestaciones de malformación linfática6).
Q¿Cuáles son los síntomas oculares del síndrome de Noonan?
La causa del NS son mutaciones de ganancia de función (o pérdida de función) en múltiples genes involucrados en la vía RAS-MAPK. Las frecuencias de los genes causales se muestran a continuación.
Gen
Frecuencia
Características principales
PTPN11
Aproximadamente 50%
Más común. Principalmente mutaciones de ganancia de función.
SOS1
Aproximadamente 10–13%
—
RAF1, RIT1
Aproximadamente 5% cada uno
Asociación reportada con discapacidad visual
LZTR1
Aproximadamente 8%
Ambos patrones de herencia AD y AR
KRAS, BRAF
Menos del 5% cada uno
Mutación BRAF asociada con informes de peor agudeza visual
LZTR1 es una proteína del aparato de Golgi perteneciente a la superfamilia BTB-Kelch que promueve la ubiquitinación y degradación de RAS, regulando negativamente la señalización RAS-MAPK 3). Las mutaciones en AD-NS se concentran en el dominio Kelch (superficie de reconocimiento del sustrato) y potencian la señalización RAS-MAPK 1)3).
En cuanto a la correlación entre genotipo y fenotipo oftálmico, se han reportado discapacidad visual permanente (mejor agudeza visual corregida < 0.3 en ambos ojos) en pacientes con mutaciones en RAF1, KRAS y SHOC2, y la peor agudeza visual (mala visión desde la infancia debido a hipoplasia del nervio óptico) en pacientes con mutaciones en BRAF. Sin embargo, no se han establecido correlaciones definitivas y las cohortes son pequeñas (n=25–105).
En el NFNS (síndrome de neurofibromatosis-Noonan) causado por mutaciones en NF1, aproximadamente el 25% de los pacientes con NF1 presentan rasgos similares a Noonan, con fenotipos superpuestos de ambas enfermedades 4).
Q¿La gravedad de los síntomas oculares varía según el tipo de gen?
A
Se ha informado de discapacidad visual permanente en pacientes con mutaciones en RAF1, KRAS y SHOC2, y de mala visión desde la infancia debido a hipoplasia del nervio óptico en pacientes con mutaciones en BRAF. Por otro lado, algunos informes indican que no se observó discapacidad visual en pacientes con mutaciones en PTPN11. Sin embargo, las cohortes que muestran estas correlaciones son pequeñas y actualmente no se ha establecido una correlación genotipo-fenotipo definitiva.
El diagnóstico de NS es evaluado por un genetista clínico según criterios diagnósticos formales basados en hallazgos clínicos como rasgos faciales característicos, enfermedad cardíaca y baja estatura.
Para el diagnóstico definitivo, se utilizan pruebas genéticas moleculares mediante un panel de genes relacionados con la vía RAS-MAPK (que incluye PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1, etc.)2)3). También se utiliza la secuenciación del exoma completo (WES)1)4). Tenga en cuenta que LZTR1 puede no estar incluido en algunos paneles de genes de NS3).
NFNS (Neurofibromatosis-Noonan syndrome): La mutación NF1 causa superposición de fenotipos de NS y NF1. Buscar comorbilidad con NF1 si hay 6 o más manchas café con leche o de tamaño ≥5 mm 4). Farncombe et al. reportaron neurofibromas plexiformes en pacientes con NS con mutación LZTR1, indicando superposición clínica con NF13).
Hipoplasia del nervio óptico: No es progresiva y la visión y el campo visual no cambian a menos que se complique con glaucoma. Evite el tratamiento de reducción de la presión intraocular innecesario. Intente mejorar la función visual residual con una corrección refractiva adecuada.
Desprendimiento seroso de retina asociado a coloboma del disco óptico: No existe un tratamiento establecido y algunos casos remiten espontáneamente. Para el desprendimiento de retina regmatógeno, considere el tratamiento quirúrgico correspondiente.
Baja estatura: Terapia con hormona de crecimiento (GH). Algunos pacientes con NS presentan deficiencia de GH 1). Sin embargo, en NFNS1, la administración de GH conlleva riesgo de aumento de neurofibromas, por lo que se requiere una evaluación cuidadosa 4).
Enfermedad cardíaca: Dilatación con balón o reparación quirúrgica para la estenosis de la válvula pulmonar. Observación y tratamiento farmacológico para la miocardiopatía hipertrófica. La miocardiopatía hipertrófica en niños menores de 2 años conlleva el mayor riesgo de muerte cardíaca 5).
Anomalías sanguíneas: Reposición de factores de coagulación. Evaluación del riesgo de sangrado preoperatorio. En el caso neonatal de Tang et al., la trombocitopenia refractaria fue el síntoma inicial, y el paciente presentaba CIA y quilotórax concomitantes 5).
Quilotórax: Drenaje torácico. En Tian et al., la microcirugía para la obstrucción de la salida del conducto torácico mejoró la hipoalbuminemia 6).
Criptorquidia: Orquidopexia en el momento adecuado.
Q¿Cómo se realiza el tratamiento ocular del síndrome de Noonan?
A
La base es la corrección refractiva con gafas o lentes de contacto y el tratamiento de la ambliopía. El estrabismo se considera para cirugía según el grado. La hipoplasia del nervio óptico no es progresiva; evite el tratamiento innecesario para reducir la presión intraocular y busque mejorar la función visual residual con una corrección refractiva adecuada. El desprendimiento seroso de retina asociado con coloboma del disco óptico puede resolverse espontáneamente y no existe un tratamiento establecido.
La fisiopatología fundamental del SN es la transducción de señales sostenida y excesiva debido a la desregulación de la vía RAS-MAPK.
PTPN11 (SHP2)
Función: Tirosina fosfatasa no receptora. Posee un mecanismo de autoinhibición mediante interacción intramolecular entre el dominio N-SH2 y el dominio PTP.
Efecto de las mutaciones NS: Se libera el mecanismo de autoinhibición, aumentando la actividad de fosfatasa (ganancia de función). La señalización RAS-MAPK se hiperactiva 2).
Diferencia con NSML: Las mutaciones de NSML (síndrome LEOPARD) son de pérdida de función en PTPN11, en contraste con NS.
LZTR1
Función: Proteína de Golgi perteneciente a la superfamilia BTB-Kelch. Promueve la poliubiquitinación y degradación proteasomal de RAS, regulando negativamente la señalización RAS-MAPK 3).
Mutaciones AD-NS: Concentradas en el dominio Kelch (superficie de reconocimiento de sustrato). Aumentan la señalización RAS-MAPK (ganancia de función) 1)3).
Mutación AR-NS: Mutación de pérdida de función. También se une al complejo RAF1/SHOC2/PP1CB y promueve la fosforilación de RAF1 Ser259 (inactivando la señalización MAPK)3).
Los componentes principales de la vía RAS-MAPK son los siguientes.
Reguladores positivos: SHP2 (PTPN11), SOS1, SOS2. Las mutaciones de ganancia de función en estos causan NS.
Promotores de la señalización RAS: MRAS, SHOC2, PPP1CB.
Cascada MAPK: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Reguladores negativos: CBL, NF1, LZTR1, SPRED1, SPRED2. Las mutaciones de pérdida de función causan aumento de la señalización3).
La vía RAS-MAPK es crucial para la diferenciación y proliferación celular. Las mutaciones de ganancia de función causan función celular anormal en tejidos sistémicos, lo que lleva a fenotipos complejos que afectan múltiples órganos.
Oftalmológicamente, la hipoplasia del nervio óptico resulta de un desarrollo deficiente de las células ganglionares de la retina y las fibras nerviosas. El coloboma del disco óptico se debe al cierre incompleto de la fisura óptica, con una prevalencia de 3 a 8 por 100,000.
En cuanto a tumores, las mutaciones de PTPN11 también se identifican como mutaciones somáticas en JMML (leucemia mielomonocítica juvenil), MDS y AML. El NS se asocia con un riesgo tres veces mayor de neoplasias hematológicas infantiles 5).
7. Investigación más reciente y perspectivas futuras (informes en etapa de investigación)
Tian et al. (2025) reportaron un caso de NS con mutación LZTR1 c.850C>T que presentaba transformación cavernosa portal, displasia del conducto torácico, linfangiectasia intestinal y enteropatía perdedora de proteínas6). Se realizó microcirugía para la obstrucción de la salida del conducto torácico, logrando la normalización de la albúmina sérica. Este hallazgo revela una nueva patología de anomalías linfáticas asociadas con NS con mutación LZTR1.
Orsolini et al. (2024) reportaron un varón de 35 años con mutación LZTR1 c.742G>A que presentaba FSH elevada y oligospermia (concentración de espermatozoides 1.5×10⁶/mL) sin antecedentes de criptorquidia7). Este caso sugiere la presencia de disfunción gonadal por daño primario de las células de Sertoli independiente de la criptorquidia, contribuyendo al esclarecimiento del mecanismo de infertilidad masculina en NS.
LZTR1 también es un gen causante de la schwannomatosis, y la investigación sobre la superposición clínica entre NS, NF1 y schwannomatosis está avanzando 3). La relación biológica entre RIT1 y LZTR1 también está atrayendo la atención, y se ha sugerido que las mutaciones en cualquiera de ellos pueden causar acumulación de RIT1 y contribuir a la sobreactivación de la vía de señalización MAPK 3).
En cuanto a las correlaciones genotipo-fenotipo, su establecimiento mediante cohortes más grandes sigue siendo un desafío futuro. Se espera la identificación de predictores del pronóstico oftalmológico y el desarrollo de dianas terapéuticas específicas para NS.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.