眼科所见

努南综合征
一目了然的要点
Section titled “一目了然的要点”1. 什么是努南综合征?
Section titled “1. 什么是努南综合征?”努南综合征(Noonan syndrome; NS)是一种由RAS-MAPK信号通路基因突变引起的遗传性疾病(RASopathy)。1968年由Jacqueline Noonan首次系统报道。
估计患病率为每1,000至2,500名新生儿中1例。遗传方式通常为常染色体显性遗传(AD),但约三分之二的病例由新生突变引起。已知LZTR1突变可表现为AD和常染色体隐性遗传(AR)两种形式;在Zhao等人报道的中国家系中,LZTR1 c.1149+1G>T突变呈AD遗传1)。
同一突变的家系内表型差异很大,即“可变表现度”,以及携带者不表现表型的“不完全外显”是其特点。Han & Park(2024)报道了一例父系遗传的PTPN11 p.Arg498Trp突变病例;父亲无症状或仅有轻度智力障碍,受累亲属的外显率估计为30–40%2)。新生突变与父亲高龄相关,认为精子发生过程中产生的突变具有选择性优势。
眼科方面,可能伴有视神经发育不全、视盘缺损、屈光不正、上睑下垂等多种异常。
Q
努南综合征的遗传概率是多少?
A
通常为常染色体显性遗传,但约三分之二的病例由新生突变引起。受累父母传给子女的理论风险为50%,但由于不完全外显,实际出现症状的比例估计为30–40%2)。有家族史者建议进行遗传咨询。
2. 主要症状和临床所见
Section titled “2. 主要症状和临床所见”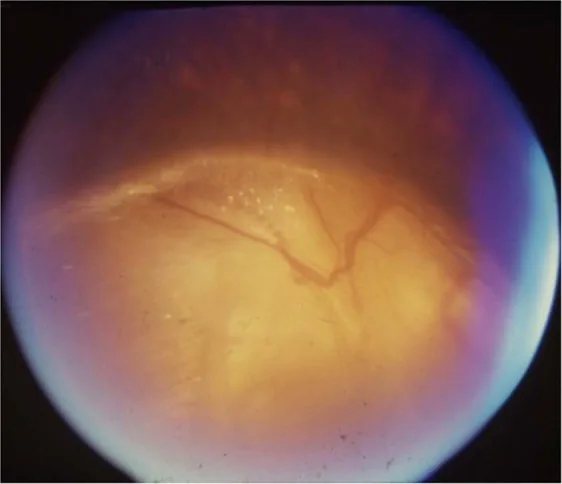
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
努南综合征的特征包括特殊面容、身材矮小和先天性心脏缺陷。
- 视力下降:由于视神经发育不全或缺损,婴儿期就可能出现视力不良。
- 斜视:自觉眼位偏斜。儿童期常由父母发现。
- 身材矮小:出生时身高正常,但随着成长,身材矮小变得明显。
- 发育迟缓:语言发育迟缓和轻度智力障碍可能是初期主诉。
全身表现
Zhao等人的病例(6.6岁女童)出现斜视、屈光不正和眼球震颤,并合并生长激素缺乏1)。Tian等人(2025)报告了LZTR1突变NS患者出现门静脉海绵样变性、肠淋巴管扩张和蛋白丢失性肠病,显示了淋巴管发育异常的多种表现6)。
Q
努南综合征的眼部症状有哪些?
3. 病因和风险因素
Section titled “3. 病因和风险因素”NS的病因是RAS-MAPK通路中多个基因的功能获得性(或功能丧失性)突变。以下列出致病基因的频率。
| 基因 | 频率 | 主要特征 |
|---|---|---|
| PTPN11 | 约50% | 最常见。主要为功能获得性突变。 |
| SOS1 | 约10~13% | — |
| RAF1, RIT1 | 各约5% | 有报道与视力障碍相关 |
| LZTR1 | 约8% | AD和AR两种遗传方式 |
| KRAS, BRAF | 各低于5% | BRAF突变有最低视力的报道 |
LZTR1是一种属于BTB-Kelch超家族的高尔基体蛋白,通过促进RAS的泛素化和降解来负调控RAS-MAPK信号3)。AD-NS的突变集中在Kelch结构域(底物识别面),从而增强RAS-MAPK信号1)3)。
关于基因型与眼科表型的相关性,有报告指出RAF1、KRAS和SHOC2突变患者出现永久性视力障碍(双眼最佳矫正视力<0.3),BRAF突变患者视力最差(因视神经发育不全导致婴儿期视力不良)。但尚未建立确定的相关性,且队列规模较小(n=25~105)。
在由NF1突变引起的NFNS(神经纤维瘤病-努南综合征)中,约25%的NF1患者表现出努南样特征,两种疾病的表型重叠4)。
Q
眼睛症状的严重程度是否因基因类型而异?
A
在RAF1、KRAS、SHOC2突变患者中已有永久性视力障碍的报道,而在BRAF突变患者中,因视神经发育不全导致婴儿期视力不良也有报道。另一方面,有报告指出PTPN11突变患者未观察到视力障碍。然而,显示这些相关性的队列规模较小,目前尚未建立确定的基因型-表型相关性。
4. 诊断与检查方法
Section titled “4. 诊断与检查方法”临床诊断与遗传学检查
Section titled “临床诊断与遗传学检查”NS的诊断由临床遗传学专家根据基于特征性面容、心脏疾病、身材矮小等临床发现的正式诊断标准进行评估。
确诊采用RAS-MAPK通路相关基因面板(包括PTPN11、SOS1、RAF1、RIT1、KRAS、LZTR1等)进行分子遗传学检测2)3)。全外显子组测序(WES)也可使用1)4)。需注意NS基因面板可能不包含LZTR13)。
诊断时建议进行全面的眼科评估,之后每年随访一次。
- 屈光检查:评估屈光不正。检查是否存在高度屈光不正和散光。
- 斜视检查:评估眼位和眼球运动。
- 眼底检查:评估视盘形态。
视神经发育不全的评估
Section titled “视神经发育不全的评估”- DM/DD比值(视盘黄斑距离/视盘直径比):≥3时怀疑视神经发育不全,≥4时可能性大。
- 双环征:视乳头周围色素环。
- 光学相干断层扫描(OCT):测量cpRNFL(视乳头周围视网膜神经纤维层)厚度有用。
- 头部MRI:评估中枢神经系统异常。约15%存在垂体漏斗异常。
视乳头缺损的诊断
Section titled “视乳头缺损的诊断”- 检眼镜所见:以下方为中心的视乳头及视网膜脉络膜缺损、血管走行异常。
- 影像检查:通过超声、MRI、CT、OCT确诊。头部MRI/CT检查有无颅内畸形。
- NFNS(神经纤维瘤病-努南综合征):NF1突变导致NS和NF1表型重叠。若存在6个以上或直径≥5mm的咖啡斑,需排查NF1合并4)。Farncombe等人报告LZTR1突变NS患者出现丛状神经纤维瘤,提示与NF1的临床重叠3)。
- 视盘缺损的鉴别:视盘周围葡萄肿、牵牛花综合征、视盘PFV/PHPV、大视盘。
- 其他综合征:CHARGE综合征、特纳综合征、心面皮肤综合征(CFC)、Costello综合征、Legius综合征。
- 超声心动图:心脏畸形筛查。
- 血液检查:凝血功能检查(PT/APTT、第XI因子活性等)5)。
5. 标准治疗方法
Section titled “5. 标准治疗方法”NS没有根治性治疗方法,以临床遗传学专家为中心的多学科协作管理是基础。
- 屈光矫正:通过眼镜或隐形眼镜矫正屈光不正。
- 弱视治疗:使用遮盖健眼等方法治疗弱视。
- 斜视手术:根据斜视程度考虑手术。
- 视神经发育不全:为非进行性,若不合并青光眼,视力和视野不会改变。避免轻易进行降眼压治疗。通过适当的屈光矫正尝试改善残余视功能。
- 视盘缺损伴发的浆液性视网膜脱离:尚无确定的治疗方法,也有自然消退的病例。对于孔源性视网膜脱离,考虑相应的手术治疗。
- 身材矮小:生长激素(GH)治疗。部分NS患者合并GH缺乏1)。但在NFNS1中,GH给药存在神经纤维瘤增大的风险,需谨慎判断4)。
- 心脏疾病:肺动脉瓣狭窄的球囊扩张术或外科修复。肥厚型心肌病的观察和药物治疗。2岁以下肥厚型心肌病患儿的心脏性死亡风险最高5)。
- 血液异常:补充凝血因子。术前出血风险评估。Tang等人的新生儿病例中,难治性血小板减少为首发症状,合并ASD和乳糜胸5)。
- 乳糜胸:胸腔引流。Tian等人通过显微手术治疗胸导管出口梗阻,改善了低蛋白血症6)。
- 隐睾:适时进行睾丸固定术。
Q
努南综合征的眼部治疗如何进行?
6. 病理生理学与详细发病机制
Section titled “6. 病理生理学与详细发病机制”NS的根本病理生理是RAS-MAPK通路调节异常导致的持续过度信号传导。
PTPN11(SHP2)
功能:非受体型酪氨酸磷酸酶。通过N-SH2结构域和PTP结构域之间的分子内相互作用具有自身抑制机制。
NS突变的影响:自身抑制机制被解除,磷酸酶活性增强(功能获得型)。RAS-MAPK信号过度激活2)。
与NSML的区别:NSML(豹皮综合征)突变是PTPN11的活性降低型,与NS相反。
LZTR1
功能:属于BTB-Kelch超家族的高尔基体蛋白。促进RAS的多聚泛素化和蛋白酶体降解,负调控RAS-MAPK信号3)。
AD-NS的突变:集中在Kelch结构域(底物识别面)。增强RAS-MAPK信号(功能获得型)1)3)。
AR-NS突变:功能丧失型突变。它还能结合RAF1/SHOC2/PP1CB复合物并促进RAF1 Ser259磷酸化(使MAPK信号失活)3)。
RAS-MAPK通路的主要组成部分如下。
- 正调控因子:SHP2(PTPN11)、SOS1、SOS2。这些因子的功能获得型突变导致NS。
- RAS信号促进因子:MRAS、SHOC2、PPP1CB。
- MAPK级联:BRAF、RAF1、MAP2K1、MAP2K2、MAPK1。
- 负调控因子:CBL、NF1、LZTR1、SPRED1、SPRED2。功能丧失型突变导致信号增强3)。
RAS-MAPK通路对细胞分化和增殖至关重要。功能获得性突变导致全身组织细胞功能异常,引起涉及多器官的复杂表型。
眼科方面,视神经发育不全由视网膜神经节细胞和神经纤维发育障碍引起。视盘缺损由眼杯裂闭合不全导致,患病率为3~8/100,000。
与肿瘤相关,PTPN11突变也被鉴定为JMML(幼年型粒单核细胞白血病)、MDS和AML的体细胞突变。NS患儿血液恶性肿瘤风险增加3倍5)。
7. 最新研究与未来展望(研究阶段报告)
Section titled “7. 最新研究与未来展望(研究阶段报告)”LZTR1突变NS中的淋巴管形成异常
Section titled “LZTR1突变NS中的淋巴管形成异常”Tian等人(2025)报告了一例LZTR1 c.850C>T突变NS病例,伴有门静脉海绵样变性、胸导管发育不全、肠淋巴管扩张和蛋白丢失性肠病6)。对胸导管出口梗阻进行了显微手术,实现了血清白蛋白的正常化。这一发现揭示了LZTR1突变NS相关淋巴管形成异常的新病理机制。
男性不育的机制
Section titled “男性不育的机制”Orsolini等人(2024)报告了一名携带LZTR1 c.742G>A突变的35岁男性,无隐睾病史,但出现高FSH和少精子症(精子浓度1.5×10⁶/mL)7)。该病例提示存在独立于隐睾的原发性支持细胞损伤导致的性腺功能障碍,有助于阐明NS中男性不育的机制。
基因型-表型相关性及未来挑战
Section titled “基因型-表型相关性及未来挑战”LZTR1也是神经鞘瘤病(schwannomatosis)的致病基因,关于NS、NF1和神经鞘瘤病临床重叠的研究正在进展中3)。RIT1与LZTR1的生物学关系也备受关注,有研究表明其中任一基因的突变可能导致RIT1积累,进而促进MAPK信号通路的过度激活3)。
关于基因型-表型相关性,通过更大规模的队列研究来确立是未来的课题。期待识别眼科预后的预测因子以及开发NS特异性的治疗靶点。
8. 参考文献
Section titled “8. 参考文献”- Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
- Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
- Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
- Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
- Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
- Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
- Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.