Синдром Нунан (Noonan syndrome; NS) — это генетическое заболевание (RASопатия), вызванное мутациями генов в сигнальном пути RAS-MAPK. Впервые систематически описан Жаклин Нунан в 1968 году.
Предполагаемая распространенность составляет 1 на 1000–2500 рождений. Наследование обычно аутосомно-доминантное (АД), но около 2/3 случаев обусловлены мутациями de novo. Известны мутации LZTR1 как с АД, так и с аутосомно-рецессивным (АР) типом наследования; в китайской семье, описанной Zhao et al., мутация LZTR1 c.1149+1G>T показала АД-наследование1).
Характерны вариабельная экспрессивность, при которой фенотип значительно различается в пределах одной семьи с одинаковой мутацией, и неполная пенетрантность, когда носители не проявляют фенотипа. Han & Park (2024) сообщили о случае отцовского наследования мутации PTPN11 p.Arg498Trp, где отец был бессимптомным или имел только легкую умственную отсталость, а частота пораженных родственников составляет 30–40%2). Мутации de novo связаны с пожилым возрастом отца, и считается, что мутации, возникающие в процессе сперматогенеза, имеют селективное преимущество.
Офтальмологически могут наблюдаться различные аномалии, включая гипоплазию зрительного нерва, колобому диска зрительного нерва, аномалии рефракции и птоз.
QКакова вероятность наследования синдрома Нунан?
A
Обычно наследование аутосомно-доминантное, но около 2/3 случаев обусловлены мутациями de novo (спонтанными). Передача от больного родителя ребенку теоретически составляет 50%, но из-за неполной пенетрантности фактическая частота проявления симптомов оценивается в 30–40%2). При наличии семейного анамнеза рекомендуется генетическое консультирование.
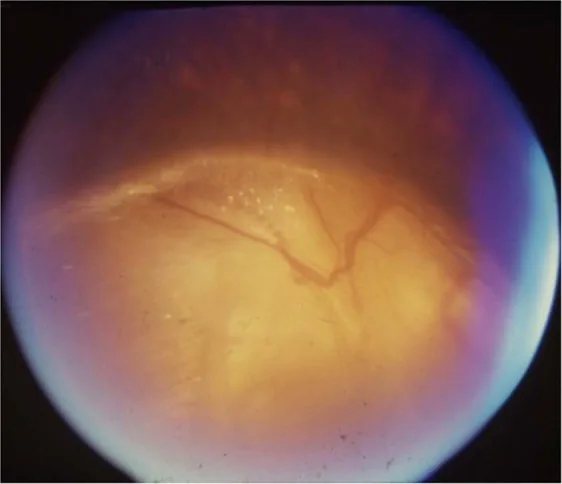
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Нейроофтальмологические признаки : косоглазие, нистагм, амблиопия, двусторонние аномалии диска зрительного нерва, включая гипоплазию, экскавацию и колобому зрительного нерва.
Системные проявления
Характерное лицо : широкий лоб, запавшая переносица, низко посаженные и ротированные кзади уши с толстой завиткой. Наиболее выражено в младенчестве и детстве.
Скелет и шея : крыловидная шея, килевидная или воронкообразная грудная клетка, широко расставленные соски, относительная макроцефалия.
Гематологические : удлинение ПВ/АЧТВ, нарушения количества и функции тромбоцитов, дефицит фактора XI (50%). Рефрактерная тромбоцитопения в неонатальном периоде может быть первым симптомом 5).
Половые органы: крипторхизм у мальчиков (до 80%), бесплодие.
В случае Zhao et al. (девочка 6,6 лет) наблюдались косоглазие, аномалии рефракции и нистагм, сочетающиеся с дефицитом гормона роста 1). Tian et al. (2025) сообщили о кавернозной трансформации воротной вены, кишечной лимфангиэктазии и протеинтеряющей энтеропатии при НС с мутацией LZTR1, что демонстрирует разнообразие лимфатических аномалий 6).
QКакие глазные симптомы наблюдаются при синдроме Нунан?
A
Наблюдаются разнообразные проявления: аномалии рефракции (миопия, гиперметропия, астигматизм), птоз, косоглазие, нистагм, амблиопия. Более тяжелые проявления включают гипоплазию зрительного нерва, колобому диска зрительного нерва и кератоконус. Рекомендуется комплексное офтальмологическое обследование с момента постановки диагноза.
Причиной НС являются мутации с усилением (или потерей) функции в нескольких генах, участвующих в пути RAS-MAPK. Частота генов-причин указана ниже.
Ген
Частота
Основные характеристики
PTPN11
Около 50%
Наиболее частый. В основном мутации с усилением функции.
SOS1
Около 10–13%
—
RAF1, RIT1
Каждый около 5%
Сообщается о связи с нарушениями зрения
LZTR1
около 8%
оба типа наследования АД и АР
KRAS, BRAF
менее 5% каждый
сообщение о наименьшей остроте зрения при мутации BRAF
LZTR1 — это белок аппарата Гольджи, принадлежащий к суперсемейству BTB-Kelch, который способствует убиквитинированию и деградации RAS, отрицательно регулируя сигнальный путь RAS-MAPK 3). Мутации при АД-НС сосредоточены в домене Kelch (поверхность распознавания субстрата) и усиливают сигнальный путь RAS-MAPK 1)3).
Что касается корреляции между генотипом и офтальмологическим фенотипом, сообщалось о постоянных нарушениях зрения (наилучшая корригированная острота зрения обоих глаз < 0,3) у пациентов с мутациями RAF1, KRAS и SHOC2, а также о минимальной остроте зрения (плохое зрение с младенчества из-за гипоплазии зрительного нерва) у пациентов с мутацией BRAF, однако окончательная корреляция не установлена, а когорты малы (n=25–105).
При NFNS (нейрофиброматоз-синдром Нунан) вследствие мутации NF1 около 25% пациентов с NF1 проявляют нунаноподобные черты, при этом фенотипы обоих заболеваний перекрываются 4).
QМеняется ли тяжесть глазных симптомов в зависимости от типа гена?
A
Сообщается о стойких нарушениях зрения у пациентов с мутациями RAF1, KRAS и SHOC2, а у пациентов с мутацией BRAF отмечается плохое зрение с младенчества из-за гипоплазии зрительного нерва. С другой стороны, есть сообщения, что у пациентов с мутацией PTPN11 нарушений зрения не наблюдалось. Однако когорты, демонстрирующие эти корреляции, невелики, и в настоящее время не установлена окончательная корреляция генотип-фенотип.
Диагноз НС устанавливается клиническим генетиком на основе формальных диагностических критериев, основанных на клинических данных, таких как характерные черты лица, заболевания сердца и низкий рост.
Для подтверждения диагноза используется молекулярно-генетическое тестирование с помощью панели генов пути RAS-MAPK (PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1 и др.)2)3). Также используется секвенирование экзома (WES)1)4). Следует отметить, что LZTR1 может не входить в генную панель НС3).
NFNS (нейрофиброматоз-синдром Нунан) : мутация NF1 приводит к перекрытию фенотипов NS+NF1. При наличии ≥6 пятен цвета кофе с молоком или размера ≥5 мм проверять на сочетание с NF14). Farncombe et al. сообщили о плексиформных нейрофибромах у пациентов с NS с мутацией LZTR1, что указывает на клиническое перекрытие с NF13).
Коррекция рефракции : коррекция аномалий рефракции с помощью очков или контактных линз.
Лечение амблиопии : лечение амблиопии с помощью окклюзии здорового глаза и т.д.
Хирургия косоглазия : Рассмотреть операцию в зависимости от степени косоглазия.
Гипоплазия зрительного нерва: непрогрессирующая, острота зрения и поле зрения не изменяются при отсутствии глаукомы. Следует избегать необоснованного снижения внутриглазного давления. Попытаться улучшить остаточную зрительную функцию с помощью соответствующей коррекции рефракции.
Серозная отслойка сетчатки, связанная с колобомой диска зрительного нерва : Стандартного лечения не существует, в некоторых случаях наблюдается спонтанное разрешение. При регматогенной отслойке сетчатки рассматривается соответствующее хирургическое лечение.
Низкий рост : терапия гормоном роста (GH). У некоторых пациентов с НС может наблюдаться дефицит GH1). Однако при НФНС1 введение GH несет риск увеличения нейрофибром, что требует осторожного решения4).
Заболевания сердца: баллонная дилатация или хирургическая коррекция стеноза клапана легочной артерии. Наблюдение и медикаментозное лечение гипертрофической кардиомиопатии. Гипертрофическая кардиомиопатия у детей младше 2 лет представляет самый высокий риск сердечной смерти5).
Нарушения крови : восполнение факторов свертывания. Оценка риска кровотечения перед операцией. В неонатальном случае Tang et al. рефрактерная тромбоцитопения была первым симптомом, сочетающимся с ДМПП и хилотораксом5).
Хилоторакс : дренирование грудной клетки. Tian et al. сообщили об улучшении гипоальбуминемии после микрохирургического лечения обструкции выхода грудного протока6).
Крипторхизм : орхидопексия в подходящее время.
QКак проводится лечение глаз при синдроме Нунан?
A
Основой являются коррекция рефракции с помощью очков или контактных линз и лечение амблиопии. Хирургическое лечение косоглазия рассматривается в зависимости от его степени. Гипоплазия зрительного нерва не прогрессирует; следует избегать необоснованного снижения внутриглазного давления и улучшать остаточную зрительную функцию с помощью адекватной коррекции рефракции. Серозная отслойка сетчатки, связанная с колобомой диска зрительного нерва, может спонтанно разрешиться, и стандартного лечения не существует.
Основная патофизиология СН — это постоянная и избыточная передача сигналов из-за нарушения регуляции пути RAS-MAPK.
PTPN11 (SHP2)
Функция: Не рецепторная тирозинфосфатаза. Имеет механизм аутоингибирования за счет внутримолекулярного взаимодействия между N-SH2 доменом и PTP доменом.
Влияние мутаций NS: Механизм аутоингибирования снимается, активность фосфатазы повышается (активирующий тип). Сигнальный путь RAS-MAPK гиперактивируется 2).
Отличие от NSML: Мутации NSML (синдром LEOPARD) являются гипоактивными формами PTPN11, в отличие от NS.
LZTR1
Функция: Белок аппарата Гольджи, принадлежащий к суперсемейству BTB-Kelch. Способствует полиубиквитинированию и протеасомной деградации RAS, отрицательно регулируя сигнальный путь RAS-MAPK 3).
Мутации AD-NS: Сосредоточены в Kelch домене (поверхность распознавания субстрата). Усиливают сигнальный путь RAS-MAPK (активирующий тип) 1)3).
Мутация AR-NS : вызвана мутацией с потерей функции. Она связывается с комплексом RAF1/SHOC2/PP1CB и способствует фосфорилированию RAF1 Ser259 (инактивация сигнала MAPK) 3).
Основные компоненты пути RAS-MAPK следующие:
Положительные регуляторы : SHP2 (PTPN11), SOS1, SOS2. Мутации с усилением функции этих генов вызывают НС.
Отрицательные регуляторы : CBL, NF1, LZTR1, SPRED1, SPRED2. Мутации с потерей функции приводят к усилению сигнала 3).
Путь RAS-MAPK крайне важен для дифференцировки и пролиферации клеток; мутации с усилением функции приводят к аномальной клеточной функции во всех тканях организма, вызывая сложный фенотип с поражением многих органов.
К офтальмологическим проявлениям относятся гипоплазия зрительного нерва, обусловленная недостаточным развитием ганглиозных клеток сетчатки и нервных волокон. Колобома диска зрительного нерва возникает из-за неполного закрытия глазной щели, распространенность составляет 3–8 на 100 000.
В отношении опухолей мутации PTPN11 также идентифицируются как соматические мутации при ЮММЛ (ювенильный миеломоноцитарный лейкоз), МДС и ОМЛ; риск гематологических злокачественных новообразований в детском возрасте при синдроме Нунан повышен в 3 раза 5).
7. Новейшие исследования и перспективы (отчеты на стадии исследований)
Tian et al. (2025) сообщили о случае НС с мутацией LZTR1 c.850C>T, проявляющемся кавернозной трансформацией воротной вены, дисплазией грудного протока, кишечной лимфангиэктазией и протеин-теряющей энтеропатией 6). Была выполнена микрохирургическая операция по устранению обструкции выхода грудного протока, что привело к нормализации уровня сывороточного альбумина. Это открытие указывает на новую патологию лимфангиогенеза, связанную с мутацией LZTR1 при НС.
Orsolini et al. (2024) сообщили о 35-летнем мужчине с мутацией LZTR1 c.742G>A, у которого наблюдались высокий уровень ФСГ и олигоспермия (концентрация сперматозоидов 1,5×10⁶/мл) без крипторхизма в анамнезе7). Этот случай предполагает наличие гонадной дисфункции вследствие первичного поражения клеток Сертоли, независимой от крипторхизма, что способствует пониманию механизма мужского бесплодия при синдроме Нунан.
Генотип-фенотипическая корреляция и будущие задачи
LZTR1 также является геном, вызывающим шванноматоз, и исследования клинического перекрытия NS, NF1 и шванноматоза продвигаются 3). Биологическая связь между RIT1 и LZTR1 также привлекает внимание, и предполагается, что мутации в любом из них могут вызывать накопление RIT1 и способствовать гиперактивации MAPK-сигнального пути 3).
Что касается корреляции генотип-фенотип, ее установление с помощью более крупных когорт является задачей на будущее. Ожидается выявление предикторов офтальмологического прогноза и разработка специфических для NS терапевтических мишеней.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.