안과적 소견

누난 증후군
한눈에 보는 포인트
섹션 제목: “한눈에 보는 포인트”1. 누난 증후군이란?
섹션 제목: “1. 누난 증후군이란?”누난 증후군(Noonan syndrome; NS)은 RAS-MAPK 신호 경로의 유전자 변이를 원인으로 하는 유전성 질환(RASopathy)입니다. 1968년 Jacqueline Noonan에 의해 처음으로 체계적으로 보고되었습니다.
추정 유병률은 출생 1,000~2,500명당 1명으로 알려져 있습니다. 유전 양식은 일반적으로 상염색체 우성 유전(AD)이지만, 약 2/3의 증례는 새로운 돌연변이(de novo)에 의한 것입니다. LZTR1 변이는 AD 및 상염색체 열성 유전(AR) 두 형태 모두 가능하며, Zhao 등의 중국 가계에서는 LZTR1 c.1149+1G>T 변이가 AD 유전을 보였습니다1).
동일한 변이를 가진 가계 내에서도 표현형이 크게 다른 ‘가변적 표현도’와 보인자가 표현형을 나타내지 않는 ‘불완전 침투’가 특징적입니다. Han & Park(2024)은 PTPN11 p.Arg498Trp 변이를 가진 부계 유전 증례를 보고했으며, 아버지는 무증상경도 지적 장애만 있었고, 발현된 친족은 3040%로 알려져 있습니다2). 새로운 돌연변이는 아버지의 고연령과 관련이 있으며, 정자 형성 과정에서 발생한 돌연변이가 선택적 우위를 가진다고 생각됩니다.
안과적으로는 시신경 저형성, 시신경 유두 결손, 굴절 이상, 안검 하수 등 다양한 이상을 동반할 수 있습니다.
Q
누난 증후군은 얼마나 유전되나요?
A
일반적으로 상염색체 우성 유전을 보이지만, 약 2/3는 새로운 돌연변이에 의한 것입니다. 이환된 부모에서 자녀로의 유전은 이론적으로 50%의 확률이지만, 불완전 침투로 인해 실제로 증상이 나타나는 비율은 30~40%로 알려져 있습니다2). 가족력이 있는 경우 유전 상담이 권장됩니다.
2. 주요 증상과 임상 소견
섹션 제목: “2. 주요 증상과 임상 소견”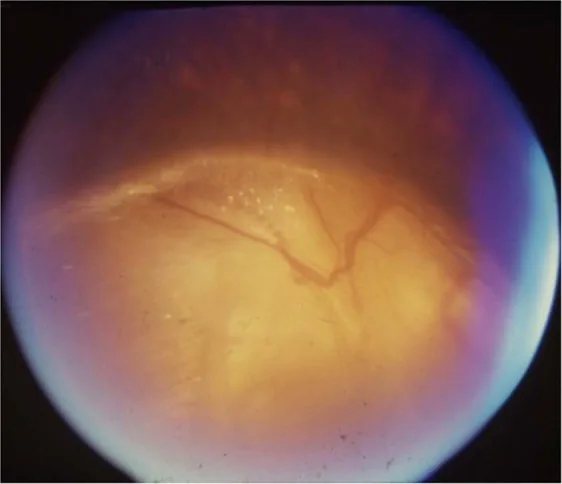
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
누난 증후군은 특징적인 얼굴 모양, 저신장, 선천성 심장 결함을 특징으로 합니다.
자각 증상
섹션 제목: “자각 증상”- 시력 저하: 시신경 저형성이나 결손으로 인해 유아기부터 시력이 나쁠 수 있습니다.
- 사시: 눈 위치의 어긋남을 자각합니다. 소아기에 부모가 알아차리는 경우가 많습니다.
- 저신장: 출생 시 키는 정상이지만 성장과 함께 저신장이 뚜렷해집니다.
- 발달 지연: 언어 발달 지연 및 경도 지적 장애가 초기 주 증상일 수 있습니다.
임상 소견
섹션 제목: “임상 소견”전신 소견
특징적 얼굴: 넓은 이마, 콧등 함몰, 두꺼운 귀 가장자리를 동반한 저위 후방 회전귀. 영아기 및 소아기에 가장 두드러짐.
심혈관계: 폐동맥 판막 협착증(50–60%), 비후성 심근병증(20%), 심방 중격 결손(6–10%), 심실 중격 결손, 림프관 형성 부전.
골격/목: 익상경, 새가슴/누두흉, 유두 간격 증가, 상대적 대두증.
혈액학적: PT/PTT 연장, 혈소판 수 및 기능 이상, 제XI인자 결핍(50%). 신생아기의 난치성 혈소판 감소증이 초기 증상일 수 있음5).
생식기: 남성의 잠복고환(최대 80%), 불임증.
Zhao 등의 증례(6.6세 여아)에서 사시, 굴절 이상, 안진이 관찰되었고, 성장호르몬 결핍을 동반하였다1). Tian 등(2025)은 LZTR1 변이 NS에서 문맥 해면상 변화, 장 림프관 확장, 단백질 소실성 장병증을 보고하였으며, 림프관 형성 이상의 다양한 병태가 제시되었다6).
Q
누난 증후군의 안과적 증상에는 어떤 것이 있습니까?
3. 원인 및 위험 요인
섹션 제목: “3. 원인 및 위험 요인”NS의 원인은 RAS-MAPK 경로에 관여하는 여러 유전자의 기능 획득(또는 기능 상실) 돌연변이이다. 원인 유전자의 빈도를 아래에 제시한다.
| 유전자 | 빈도 | 주요 특징 |
|---|---|---|
| PTPN11 | 약 50% | 가장 흔함. 주로 기능 획득 돌연변이. |
| SOS1 | 약 10~13% | — |
| RAF1, RIT1 | 각 약 5% | 시력 장애와의 연관성 보고됨 |
| LZTR1 | 약 8% | AD 및 AR 두 유전 형태 |
| KRAS, BRAF | 각각 5% 미만 | BRAF 돌연변이에서 최저 시력 보고 |
LZTR1은 BTB-Kelch 슈퍼패밀리에 속하는 골지체 단백질로, RAS의 유비퀴틴화 및 분해를 촉진하여 RAS-MAPK 신호를 음성적으로 조절합니다3). AD-NS의 돌연변이는 Kelch 도메인(기질 인식면)에 집중되어 RAS-MAPK 신호를 증가시킵니다1)3).
유전자형과 안과적 표현형의 상관관계에 대해, RAF1, KRAS, SHOC2 돌연변이 환자에서 영구적 시력 장애(양안 최대교정시력 < 0.3), BRAF 돌연변이 환자에서 최저 시력(시신경 저형성으로 인한 유아기부터의 시력 불량)이 보고되었으나, 확정적인 상관관계는 확립되지 않았으며 코호트 규모가 작습니다(n=25~105).
NF1 돌연변이로 인한 NFNS(신경섬유종증-누난 증후군)에서는 NF1 환자의 약 25%가 누난 유사 특징을 보이며, 두 질환의 표현형이 중복됩니다4).
Q
유전자 종류에 따라 눈 증상의 심각도가 달라지나요?
A
RAF1, KRAS, SHOC2 돌연변이 환자에서 영구적인 시력 장애가 보고되었으며, BRAF 돌연변이 환자에서는 시신경 저형성으로 인한 유아기부터의 시력 저하가 보고되었습니다. 반면, PTPN11 돌연변이 환자에서는 시력 장애가 관찰되지 않았다는 보고도 있습니다. 그러나 이러한 상관관계를 보여주는 코호트는 규모가 작으며, 현재 확정적인 유전자형-표현형 상관관계는 확립되지 않았습니다.
4. 진단 및 검사 방법
섹션 제목: “4. 진단 및 검사 방법”임상 진단 및 유전학적 검사
섹션 제목: “임상 진단 및 유전학적 검사”NS의 진단은 특징적인 얼굴 모습, 심장 질환, 저신장 등의 임상 소견에 기반한 공식 진단 기준에 따라 임상 유전 전문의가 평가합니다.
확진을 위해 RAS-MAPK 경로 관련 유전자 패널(PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1 등)을 이용한 분자 유전학적 검사가 사용됩니다2)3). 전장 엑솜 시퀀싱(WES)도 사용됩니다1)4). NS 유전자 패널에 LZTR1이 포함되지 않은 경우가 있을 수 있으므로 주의가 필요합니다3).
안과 검사
섹션 제목: “안과 검사”진단 시 포괄적인 안과 평가를 시행하고, 이후 매년 추적 관찰이 권장됩니다.
- 굴절 검사: 굴절 이상 평가. 고도 굴절 이상 및 난시 유무 확인.
- 사시 검사: 안위 및 안구 운동 평가.
- 안저 검사: 시신경 유두 형태 평가.
시신경 형성 부전 평가
섹션 제목: “시신경 형성 부전 평가”- DM/DD 비율(유두황반거리/유두직경비): 3 이상이면 시신경 형성 부전 의심, 4 이상이면 가능성 높음.
- 이중 고리 징후(double ring sign): 유두 주위 색소 고리.
- 광간섭단층촬영(OCT): cpRNFL(유두 주위 망막신경섬유층) 두께 측정이 유용합니다.
- 머리 MRI: 중추신경계 이상 평가. 약 15%에서 뇌하수체 누두부 이상이 관찰됩니다.
시신경 유두 결손의 진단
섹션 제목: “시신경 유두 결손의 진단”- 검안경 소견: 아래쪽을 중심으로 한 유두 및 망막맥락막 결손, 혈관 주행 이상.
- 영상 검사: 초음파, MRI, CT, OCT를 통한 확진. 머리 MRI/CT로 두개내 기형 동반 여부 확인.
감별 진단
섹션 제목: “감별 진단”- NFNS(신경섬유종증-누난 증후군): NF1 돌연변이로 NS와 NF1 표현형이 중복됨. 6개 이상 또는 크기 5mm 이상의 카페오레 반점이 있으면 NF1 동반을 검사합니다4). Farncombe 등은 LZTR1 돌연변이 NS 환자에서 총상 신경섬유종을 발견하고 NF1과의 임상적 중복을 보고했습니다3).
- 시신경 유두 결손의 감별: 유두주위 포도종, 모닝글로리 증후군, 유두부 PFV/PHPV, 거대 유두증.
- 기타 증후군: CHARGE 증후군, 터너 증후군, 심안면피부 증후군(CFC), 코스텔로 증후군, 레지우스 증후군.
기타 검사
섹션 제목: “기타 검사”- 심초음파: 심장 기형 선별검사.
- 혈액 검사: 응고 기능 검사(PT/APTT, 제XI인자 활성 등) 5).
5. 표준 치료법
섹션 제목: “5. 표준 치료법”NS는 근치적 치료법이 없으며, 임상 유전 전문의를 중심으로 한 다학제 협력에 의한 종합적 관리가 기본입니다.
안과적 관리
섹션 제목: “안과적 관리”- 굴절 교정: 굴절 이상에 대한 안경 또는 콘택트렌즈를 통한 교정.
- 약시 치료: 건안 가림 등을 이용한 약시 치료.
- 사시 수술: 사시 정도에 따라 수술을 고려합니다.
- 시신경 형성부전: 비진행성이며, 녹내장이 합병되지 않으면 시력과 시야는 변하지 않습니다. 경솔한 안압 하강 치료는 삼가합니다. 적절한 굴절 교정으로 잔존 시기능 개선을 시도합니다.
- 시신경 유두 결손에 동반된 장액성 망막박리: 정해진 치료법은 없으며, 자연 소실되는 경우도 있습니다. 열공성 망막박리에는 이에 준한 수술 요법을 고려합니다.
전신 관리
섹션 제목: “전신 관리”- 저신장: 성장호르몬(GH) 요법. 일부 NS 환자에서 GH 결핍이 동반됩니다1). 그러나 NFNS1에서는 GH 투여로 신경섬유종 증가 위험이 있어 신중한 판단이 필요합니다4).
- 심장 질환: 폐동맥판 협착증에 대한 풍선 확장술 또는 외과적 교정. 비후성 심근병증의 경과 관찰 및 약물 치료. 2세 미만의 비후성 심근병증은 심장사 최고 위험입니다5).
- 혈액 이상:응고인자 보충. 수술 전 출혈 위험 평가. Tang 등의 신생아 증례에서는 불응성 혈소판 감소증이 초기 증상이었으며, ASD와 유미흉이 동반되었습니다 5).
- 유미흉:흉강 배액. Tian 등은 흉관 출구 폐쇄에 대한 현미경 수술로 저알부민혈증이 개선되었습니다 6).
- 잠복고환:적절한 시기에 고환 고정술.
Q
누난 증후군의 눈 치료는 어떻게 이루어지나요?
6. 병태생리학 및 상세 발병 기전
섹션 제목: “6. 병태생리학 및 상세 발병 기전”NS의 근본적인 병태생리는 RAS-MAPK 경로의 조절 이상으로 인한 지속적이고 과도한 신호 전달입니다.
PTPN11 (SHP2)
기능: 비수용체형 티로신 포스파타제. N-SH2 도메인과 PTP 도메인 간의 분자 내 상호작용을 통한 자가억제 기전을 가짐.
NS 돌연변이의 영향: 자가억제 기전이 해제되어 포스파타제 활성이 항진됨(기능획득형). RAS-MAPK 신호가 과활성화됨 2).
NSML과의 차이: NSML(레오파드 증후군) 돌연변이는 PTPN11의 활성 저하형으로, NS와 대조적임.
LZTR1
기능: BTB-Kelch 슈퍼패밀리에 속하는 골지체 단백질. RAS의 폴리유비퀴틴화 및 프로테아좀 분해를 촉진하여 RAS-MAPK 신호를 음성 조절함 3).
AD-NS의 돌연변이: Kelch 도메인(기질 인식면)에 집중됨. RAS-MAPK 신호를 항진시킴(기능획득형) 1)3).
AR-NS 돌연변이: 기능 상실형 돌연변이. 또한 RAF1/SHOC2/PP1CB 복합체에 결합하여 RAF1 Ser259 인산화를 촉진합니다(MAPK 신호 비활성화)3).
RAS-MAPK 경로의 주요 구성 요소는 다음과 같습니다.
- 양성 조절 인자: SHP2(PTPN11), SOS1, SOS2. 이러한 유전자의 기능 획득형 돌연변이가 NS의 원인이 됩니다.
- RAS 신호 촉진 인자: MRAS, SHOC2, PPP1CB.
- MAPK 캐스케이드: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
- 음성 조절 인자: CBL, NF1, LZTR1, SPRED1, SPRED2. 기능 상실형 돌연변이가 신호 항진의 원인이 됩니다3).
RAS-MAPK 경로는 세포 분화와 증식에 매우 중요합니다. 기능 획득 돌연변이로 인해 전신 조직에서 비정상적인 세포 기능이 발생하여 여러 장기에 걸친 복잡한 표현형을 초래합니다.
안과적 병태로는 시신경 저형성이 망막 신경절 세포 및 신경 섬유의 발생 부전으로 인해 발생합니다. 시신경 유두 결손은 안배열의 폐쇄 부전으로 인해 발생하며, 유병률은 3~8/100,000으로 알려져 있습니다.
종양과의 관련성으로, PTPN11 돌연변이는 JMML(소아 골수단구성 백혈병), MDS, AML의 체세포 돌연변이로도 확인되었으며, NS는 소아기 혈액 악성 종양 위험이 3배로 알려져 있습니다5).
7. 최신 연구와 향후 전망 (연구 단계 보고)
섹션 제목: “7. 최신 연구와 향후 전망 (연구 단계 보고)”LZTR1 변이 NS에서의 림프관 형성 이상
섹션 제목: “LZTR1 변이 NS에서의 림프관 형성 이상”Tian 등(2025)은 LZTR1 c.850C>T 변이 NS 증례에서 문맥 해면상 변성, 흉관 형성 부전, 장 림프관 확장증, 단백질 소실성 장병증을 보고했습니다6). 흉관 출구 폐쇄에 대한 현미경 수술을 시행하여 혈청 알부민 정상화를 달성했습니다. 이는 LZTR1 변이 NS에 동반된 림프관 형성 이상의 새로운 병태를 보여주는 소견입니다.
남성 불임의 기전
섹션 제목: “남성 불임의 기전”Orsolini 등(2024)은 LZTR1 c.742G>A 변이를 가진 35세 남성에서 잠복고환 병력 없이 고FSH 및 희소정자증(정자 농도 1.5×10⁶/mL)을 확인했다고 보고했습니다7). 이 증례는 잠복고환과 독립적인 원발성 Sertoli 세포 손상에 의한 성선 기능 장애의 존재를 시사하며, NS에서 남성 불임의 기전 규명에 기여하는 소견입니다.
유전자형-표현형 상관관계 및 향후 과제
섹션 제목: “유전자형-표현형 상관관계 및 향후 과제”LZTR1은 신경초종증(schwannomatosis)의 원인 유전자이기도 하며, NS, NF1 및 신경초종증 간의 임상적 중복에 대한 연구가 진행 중입니다3). RIT1과 LZTR1의 생물학적 관계에도 주목하고 있으며, 둘 중 하나의 돌연변이가 RIT1 축적을 유발하여 MAPK 신호의 과활성화에 기여할 가능성이 제시되었습니다3).
유전자형-표현형 상관관계에 대해서는 더 큰 규모의 코호트를 통한 확립이 향후 과제입니다. 안과적 예후 예측 인자의 동정 및 NS 특이적 치료 표적의 개발이 기대됩니다.
8. 참고문헌
섹션 제목: “8. 참고문헌”- Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
- Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
- Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
- Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
- Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
- Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
- Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.