La sindrome di Noonan (NS) è una malattia genetica (RASopatia) causata da mutazioni genetiche nella via di segnalazione RAS-MAPK. Fu descritta per la prima volta in modo sistematico da Jacqueline Noonan nel 1968.
La prevalenza stimata è di 1 su 1.000-2.500 nati. La modalità di trasmissione è solitamente autosomica dominante (AD), ma circa 2/3 dei casi sono dovuti a mutazioni de novo. Sono note mutazioni LZTR1 sia in forma AD che autosomica recessiva (AR); in una famiglia cinese riportata da Zhao et al., la mutazione LZTR1 c.1149+1G>T ha mostrato una trasmissione AD1).
Sono caratteristiche l’espressività variabile, per cui il fenotipo differisce notevolmente all’interno della stessa famiglia con la stessa mutazione, e la penetranza incompleta, per cui i portatori non mostrano il fenotipo. Han & Park (2024) hanno riportato un caso di trasmissione paterna della mutazione PTPN11 p.Arg498Trp, in cui il padre era asintomatico o presentava solo un lieve deficit intellettivo, e la percentuale di parenti affetti è stimata al 30-40%2). Le mutazioni de novo sono associate a un’età paterna avanzata e si ritiene che le mutazioni insorte durante la spermatogenesi abbiano un vantaggio selettivo.
Dal punto di vista oftalmologico, possono essere associate varie anomalie, tra cui ipoplasia del nervo ottico, coloboma della papilla ottica, errori di rifrazione e ptosi.
QQuanto è ereditabile la sindrome di Noonan?
A
La trasmissione è solitamente autosomica dominante, ma circa 2/3 dei casi sono dovuti a mutazioni de novo (spontanee). La trasmissione da un genitore affetto a un figlio è teoricamente del 50%, ma a causa della penetranza incompleta, la percentuale effettiva di manifestazione dei sintomi è stimata al 30-40%2). In caso di storia familiare, si raccomanda la consulenza genetica.
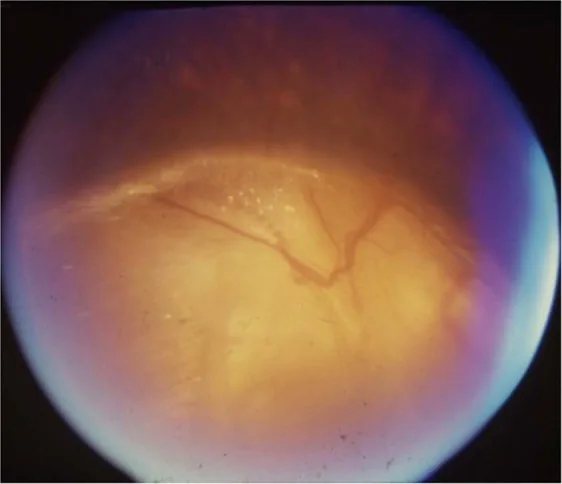
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Reperti neuro-oftalmologici : strabismo, nistagmo, ambliopia, anomalie bilaterali della papilla ottica inclusi ipoplasia, escavazione e coloboma del nervo ottico.
Reperti sistemici
Facies caratteristica : fronte ampia, radice del naso infossata, orecchie basse e ruotate posteriormente con elice spesso. Più evidente nell’infanzia e nella fanciullezza.
Sistema cardiovascolare : stenosi della valvola polmonare (50-60%), cardiomiopatia ipertrofica (20%), difetto del setto interatriale (6-10%), difetto del setto interventricolare, displasia linfatica.
Scheletro e collo : pterigio colli, pectus carinatum o excavatum, ipertelorismo mammellare, macrocefalia relativa.
Reperti ematologici : allungamento del PT/PTT, anomalie del numero e della funzione piastrinica, deficit del fattore XI (50%). La trombocitopenia refrattaria neonatale può essere il sintomo d’esordio 5).
Genitali: criptorchidismo nei maschi (fino all’80%), infertilità.
Nel caso di Zhao et al. (bambina di 6,6 anni) sono stati osservati strabismo, errori refrattivi e nistagmo, associati a deficit dell’ormone della crescita 1). Tian et al. (2025) hanno riportato trasformazione cavernosa della vena porta, linfangiectasia intestinale ed enteropatia proteinodisperdente nella NS con mutazione LZTR1, dimostrando la varietà delle anomalie linfatiche 6).
QQuali sono i sintomi oculari della sindrome di Noonan?
La NS è causata da mutazioni gain-of-function (o loss-of-function) in diversi geni coinvolti nella via RAS-MAPK. La frequenza dei geni causali è indicata di seguito.
Gene
Frequenza
Caratteristiche principali
PTPN11
Circa il 50%
Più comune. Mutazioni gain-of-function principalmente.
SOS1
Circa il 10-13%
—
RAF1, RIT1
Ciascuno circa il 5%
Segnalata associazione con disturbi visivi
LZTR1
circa 8%
ereditarietà AD e AR
KRAS, BRAF
ciascuno <5%
segnalazione di minima acuità visiva con mutazione BRAF
LZTR1 è una proteina dell’apparato di Golgi appartenente alla superfamiglia BTB-Kelch, che promuove l’ubiquitinazione e la degradazione di RAS, regolando negativamente il segnale RAS-MAPK 3). Le mutazioni della NS-AD si concentrano nel dominio Kelch (superficie di riconoscimento del substrato) e aumentano il segnale RAS-MAPK 1)3).
Per quanto riguarda la correlazione tra genotipo e fenotipo oftalmico, sono state riportate disabilità visive permanenti (migliore acuità visiva corretta binoculare < 0,3) in pazienti con mutazioni RAF1, KRAS e SHOC2, e acuità visiva minima (scarsa visione fin dall’infanzia a causa di ipoplasia del nervo ottico) in pazienti con mutazione BRAF, ma una correlazione definitiva non è stabilita e le coorti sono di piccole dimensioni (n=25-105).
Nella NFNS (neurofibromatosi-sindrome di Noonan) dovuta a mutazione NF1, circa il 25% dei pazienti NF1 mostra caratteristiche simil-Noonan, con sovrapposizione dei fenotipi di entrambe le malattie 4).
QLa gravità dei sintomi oculari varia in base al tipo di gene?
A
Sono state riportate menomazioni visive permanenti in pazienti con mutazioni RAF1, KRAS e SHOC2, e una scarsa visione fin dall’infanzia dovuta a ipoplasia del nervo ottico è stata segnalata in pazienti con mutazione BRAF. D’altra parte, alcuni studi non hanno riscontrato menomazioni visive in pazienti con mutazione PTPN11. Tuttavia, le coorti che mostrano queste correlazioni sono di piccole dimensioni e al momento non è stata stabilita una correlazione genotipo-fenotipo definitiva.
La diagnosi di NS viene valutata da un genetista clinico secondo criteri diagnostici formali basati su reperti clinici come aspetto facciale caratteristico, cardiopatie e bassa statura.
Per la diagnosi di conferma si utilizza il test genetico molecolare tramite pannello genico della via RAS-MAPK (PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1, ecc.)2)3). Viene anche utilizzato il sequenziamento dell’esoma (WES)1)4). Da notare che LZTR1 potrebbe non essere incluso nel pannello genico per NS3).
NFNS (neurofibromatosi-sindrome di Noonan) : mutazione NF1 causa sovrapposizione dei fenotipi NS+NF1. Se ≥6 macchie caffè-latte o dimensioni ≥5 mm, ricercare co-occorrenza di NF14). Farncombe et al. hanno riportato neurofibromi plessiformi in pazienti NS con mutazione LZTR1, mostrando sovrapposizione clinica con NF13).
Correzione refrattiva : correzione dei vizi di refrazione con occhiali o lenti a contatto.
Trattamento dell’ambliopia : trattamento dell’ambliopia mediante occlusione dell’occhio sano, ecc.
Chirurgia dello strabismo : prendere in considerazione l’intervento chirurgico in base al grado di strabismo.
Ipoplasia del nervo ottico: non progressiva, la vista e il campo visivo non cambiano se non si associa glaucoma. Evitare trattamenti ipotonizzanti oculari non necessari. Tentare di migliorare la funzione visiva residua con un’adeguata correzione refrattiva.
Distacco sieroso di retina associato a coloboma del disco ottico : Non esiste un trattamento standardizzato e in alcuni casi si verifica una risoluzione spontanea. Per il distacco regmatogeno di retina si considera un intervento chirurgico appropriato.
Bassa statura : terapia con ormone della crescita (GH). Alcuni pazienti con NS presentano deficit di GH1). Tuttavia, nella NFNS1 la somministrazione di GH comporta un rischio di aumento dei neurofibromi, richiedendo una decisione attenta4).
Cardiopatia: dilatazione con palloncino o riparazione chirurgica per la stenosi della valvola polmonare. Monitoraggio e trattamento farmacologico della cardiomiopatia ipertrofica. La cardiomiopatia ipertrofica nei bambini di età inferiore a 2 anni rappresenta il più alto rischio di morte cardiaca5).
Anomalie del sangue : supplementazione di fattori della coagulazione. Valutazione del rischio emorragico pre-operatorio. Nel caso neonatale di Tang et al., la trombocitopenia refrattaria è stata il primo sintomo, associata a CIA e chilotorace5).
Chilotorace : drenaggio toracico. Tian et al. hanno riportato un miglioramento dell’ipoalbuminemia dopo microchirurgia per ostruzione dello sbocco del dotto toracico6).
Criptorchidismo : orchidopessi al momento opportuno.
QCome viene trattato l'occhio nella sindrome di Noonan?
A
La correzione refrattiva con occhiali o lenti a contatto e il trattamento dell’ambliopia sono la base. Lo strabismo viene considerato per la chirurgia in base al grado. L’ipoplasia del nervo ottico non è progressiva; evitare un trattamento ipotonizzante inappropriato e migliorare la funzione visiva residua con un’adeguata correzione refrattiva. Il distacco sieroso di retina associato al coloboma del disco ottico può regredire spontaneamente e non esiste un trattamento standardizzato.
6. Fisiopatologia e meccanismi patogenetici dettagliati
La fisiopatologia di base della SN è una segnalazione persistente ed eccessiva dovuta a disregolazione della via RAS-MAPK.
PTPN11 (SHP2)
Funzione: Tirosina fosfatasi non recettoriale. Possiede un meccanismo di autoinibizione tramite interazione intramolecolare tra il dominio N-SH2 e il dominio PTP.
Effetto delle mutazioni NS: Il meccanismo di autoinibizione viene rimosso, l’attività fosfatasica è aumentata (guadagno di funzione). La segnalazione RAS-MAPK è iperattivata 2).
Differenza con NSML: Le mutazioni NSML (sindrome LEOPARD) sono forme ipoatte di PTPN11, al contrario di NS.
LZTR1
Funzione: Proteina dell’apparato di Golgi appartenente alla superfamiglia BTB-Kelch. Promuove la poliubiquitinazione e la degradazione proteasomica di RAS, regolando negativamente la segnalazione RAS-MAPK 3).
Mutazioni AD-NS: Concentrate nel dominio Kelch (superficie di riconoscimento del substrato). Aumentano la segnalazione RAS-MAPK (guadagno di funzione) 1)3).
Mutazione AR-NS : dovuta a mutazione con perdita di funzione. Si lega al complesso RAF1/SHOC2/PP1CB e promuove la fosforilazione di RAF1 Ser259 (inattivazione del segnale MAPK) 3).
I principali componenti della via RAS-MAPK sono:
Regolatori positivi : SHP2 (PTPN11), SOS1, SOS2. Mutazioni gain-of-function di questi geni causano NS.
Fattori di promozione del segnale RAS : MRAS, SHOC2, PPP1CB.
Cascata MAPK : BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Regolatori negativi : CBL, NF1, LZTR1, SPRED1, SPRED2. Mutazioni con perdita di funzione causano un aumento del segnale 3).
La via RAS-MAPK è fondamentale per la differenziazione e proliferazione cellulare; mutazioni gain-of-function causano una funzione cellulare anomala in tutti i tessuti dell’organismo, determinando un fenotipo complesso che coinvolge più organi.
Dal punto di vista oftalmologico, l’ipoplasia del nervo ottico è dovuta a uno sviluppo insufficiente delle cellule gangliari retiniche e delle fibre nervose. Il coloboma del disco ottico deriva da una chiusura incompleta della fessura ottica, con una prevalenza stimata di 3-8/100.000.
Per quanto riguarda i tumori, le mutazioni di PTPN11 sono identificate anche come mutazioni somatiche in JMML (leucemia mielomonocitica giovanile), MDS e LMA; il rischio di neoplasie ematologiche infantili nella sindrome di Noonan è triplicato 5).
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Tian et al. (2025) hanno riportato un caso di NS con mutazione LZTR1 c.850C>T che presentava trasformazione cavernomatosa della vena porta, aplasia del dotto toracico, linfangiectasia intestinale ed enteropatia proteinodisperdente 6). È stato eseguito un intervento microchirurgico per l’ostruzione dello sbocco del dotto toracico, con normalizzazione dell’albumina sierica. Ciò evidenzia una nuova patologia della linfangiogenesi associata alla mutazione LZTR1 nel NS.
Orsolini et al. (2024) hanno riportato un uomo di 35 anni con mutazione LZTR1 c.742G>A che presentava FSH elevato e oligospermia (concentrazione di spermatozoi 1,5×10⁶/mL) senza precedenti di criptorchidismo7). Questo caso suggerisce l’esistenza di una disfunzione gonadica dovuta a un danno primario delle cellule di Sertoli indipendente dal criptorchidismo, contribuendo alla comprensione del meccanismo dell’infertilità maschile nella sindrome di Noonan.
LZTR1 è anche un gene causativo della schwannomatosi, e la ricerca sulla sovrapposizione clinica tra NS, NF1 e schwannomatosi sta progredendo 3). Anche la relazione biologica tra RIT1 e LZTR1 sta attirando attenzione, ed è stato suggerito che mutazioni in uno dei due potrebbero causare accumulo di RIT1 e contribuire all’iperattivazione del segnale MAPK 3).
Per quanto riguarda la correlazione genotipo-fenotipo, la sua definizione tramite coorti più ampie è una sfida futura. L’identificazione di fattori predittivi della prognosi oftalmologica e lo sviluppo di bersagli terapeutici specifici per NS sono attesi.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.