Le syndrome de Noonan (SN) est une maladie génétique (RASopathie) causée par des mutations génétiques dans la voie de signalisation RAS-MAPK. Il a été décrit pour la première fois de manière systématique par Jacqueline Noonan en 1968.
La prévalence estimée est de 1 pour 1 000 à 2 500 naissances. Le mode de transmission est généralement autosomique dominant (AD), mais environ 2/3 des cas sont dus à des mutations de novo. Des mutations LZTR1, à la fois AD et autosomiques récessives (AR), sont connues ; dans une famille chinoise rapportée par Zhao et al., la mutation LZTR1 c.1149+1G>T a montré une transmission AD1).
L’expressivité variable, où le phénotype diffère considérablement au sein d’une même famille portant la même mutation, et la pénétrance incomplète, où les porteurs ne présentent pas de phénotype, sont caractéristiques. Han & Park (2024) ont rapporté un cas de transmission paternelle de la mutation PTPN11 p.Arg498Trp, où le père était asymptomatique ou présentait seulement une déficience intellectuelle légère, et 30 à 40 % des parents affectés sont rapportés2). Les mutations de novo sont associées à un âge paternel avancé, et on pense que les mutations survenant pendant la spermatogenèse ont un avantage sélectif.
QQuelle est la probabilité de transmission héréditaire du syndrome de Noonan ?
A
La transmission est généralement autosomique dominante, mais environ 2/3 des cas sont dus à des mutations de novo (spontanées). La transmission d’un parent atteint à un enfant est théoriquement de 50 %, mais en raison d’une pénétrance incomplète, le taux réel de manifestation des symptômes est estimé à 30-40 %2). Un conseil génétique est recommandé en cas d’antécédents familiaux.
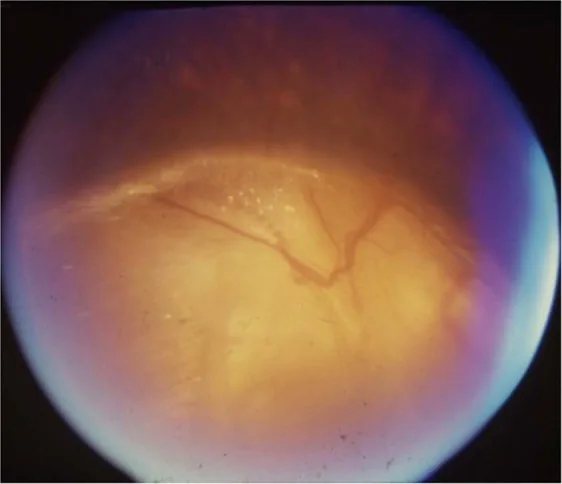
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Signes neuro-ophtalmologiques : strabisme, nystagmus, amblyopie, anomalies bilatérales de la papille optique incluant hypoplasie, excavation et colobome du nerf optique.
Signes généraux
Facies caractéristique : front large, dépression de la racine nasale, oreilles basses et postérieurement rotées avec un hélix épais. Plus marqué pendant la période infantile et l’enfance.
Système cardiovasculaire : sténose de la valve pulmonaire (50-60%), cardiomyopathie hypertrophique (20%), communication interauriculaire (6-10%), communication interventriculaire, dysplasie lymphatique.
Squelette et cou : pterygium colli, thorax en carène ou en entonnoir, hypertélorisme mamelonnaire, macrocéphalie relative.
Hématologie : allongement du TP/TCA, anomalies du nombre et de la fonction plaquettaires, déficit en facteur XI (50%). Une thrombopénie réfractaire néonatale peut être le premier symptôme 5).
Organes génitaux : cryptorchidie chez les garçons (jusqu’à 80 %), infertilité.
Dans le cas de Zhao et al. (fille de 6,6 ans), on a observé un strabisme, des erreurs de réfraction et un nystagmus, associés à un déficit en hormone de croissance 1). Tian et al. (2025) ont rapporté une transformation caverneuse de la veine porte, une lymphangiectasie intestinale et une entéropathie avec perte de protéines dans le syndrome de Noonan avec mutation LZTR1, montrant la diversité des anomalies lymphatiques 6).
QQuels sont les symptômes oculaires du syndrome de Noonan ?
Le syndrome de Noonan est causé par des mutations gain-de-fonction (ou perte-de-fonction) de plusieurs gènes impliqués dans la voie RAS-MAPK. La fréquence des gènes responsables est indiquée ci-dessous.
Gène
Fréquence
Principales caractéristiques
PTPN11
Environ 50 %
Le plus fréquent. Mutations gain-de-fonction principalement.
SOS1
Environ 10 à 13 %
—
RAF1, RIT1
Environ 5 % chacun
Association rapportée avec des troubles visuels
LZTR1
environ 8%
transmission AD et AR
KRAS, BRAF
moins de 5% chacun
rapport de la plus faible acuité visuelle avec mutation BRAF
LZTR1 est une protéine de l’appareil de Golgi appartenant à la superfamille BTB-Kelch, qui favorise l’ubiquitination et la dégradation de RAS, régulant négativement la signalisation RAS-MAPK 3). Les mutations de NS-AD se concentrent dans le domaine Kelch (surface de reconnaissance du substrat) et augmentent la signalisation RAS-MAPK 1)3).
Concernant la corrélation entre le génotype et le phénotype ophtalmique, des déficiences visuelles permanentes (meilleure acuité visuelle corrigée binoculaire < 0,3) ont été rapportées chez les patients présentant des mutations RAF1, KRAS et SHOC2, et une acuité visuelle minimale (mauvaise vision depuis la petite enfance due à une hypoplasie du nerf optique) chez les patients avec mutation BRAF, mais une corrélation définitive n’est pas établie et les cohortes sont de petite taille (n=25 à 105).
Dans le NFNS (neurofibromatose de type 1 - syndrome de Noonan) dû à une mutation NF1, environ 25 % des patients NF1 présentent des caractéristiques de type Noonan, avec un chevauchement des phénotypes des deux maladies 4).
QLa gravité des symptômes oculaires varie-t-elle selon le type de gène ?
A
Des rapports de déficience visuelle permanente existent chez les patients présentant des mutations RAF1, KRAS et SHOC2, et une mauvaise vision depuis la petite enfance due à une hypoplasie du nerf optique a été rapportée chez les patients avec mutation BRAF. En revanche, certaines études n’ont pas observé de déficience visuelle chez les patients avec mutation PTPN11. Cependant, les cohortes montrant ces corrélations sont de petite taille et aucune corrélation génotype-phénotype définitive n’est établie à ce jour.
Le diagnostic du NS est évalué par un généticien clinicien selon des critères diagnostiques formels basés sur des signes cliniques tels que la dysmorphie faciale, les cardiopathies et la petite taille.
Pour un diagnostic de confirmation, on utilise des tests de génétique moléculaire par panel de gènes de la voie RAS-MAPK (PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1, etc.)2)3). Le séquençage de l’exome entier (WES) est également utilisé1)4). Il faut noter que LZTR1 peut ne pas être inclus dans le panel génétique du NS3).
NFNS (neurofibromatose-syndrome de Noonan) : mutation NF1 entraînant un chevauchement des phénotypes NS+NF1. Rechercher une association NF1 si ≥6 taches café-au-lait ou de taille ≥5 mm 4). Farncombe et al. ont rapporté des neurofibromes plexiformes chez des patients NS avec mutation LZTR1, montrant un chevauchement clinique avec NF13).
Correction réfractive : correction des anomalies de réfraction par lunettes ou lentilles de contact.
Traitement de l’amblyopie : traitement de l’amblyopie par occlusion de l’œil sain, etc.
Chirurgie du strabisme : envisager une intervention chirurgicale en fonction du degré de strabisme.
Hypoplasie du nerf optique : non progressive, la vision et le champ visuel ne changent pas en l’absence de glaucome. Éviter un traitement abaissant la pression intraoculaire inutile. Tenter d’améliorer la fonction visuelle résiduelle par une correction réfractive appropriée.
Décollement séreux de la rétine associé à un colobome de la papille optique : Il n’existe pas de traitement standardisé et des cas de régression spontanée sont observés. Pour le décollement rhegmatogène de la rétine, une chirurgie adaptée est envisagée.
Petite taille : traitement par hormone de croissance (GH). Certains patients atteints de NS présentent un déficit en GH1). Cependant, dans le NFNS1, l’administration de GH comporte un risque d’augmentation des neurofibromes, nécessitant une décision prudente4).
Cardiopathie : dilatation par ballonnet ou réparation chirurgicale pour la sténose de la valve pulmonaire. Surveillance et traitement médicamenteux de la cardiomyopathie hypertrophique. La cardiomyopathie hypertrophique chez les enfants de moins de 2 ans présente le risque le plus élevé de mort cardiaque5).
Anomalies sanguines : apport de facteurs de coagulation. Évaluation du risque hémorragique avant chirurgie. Dans le cas néonatal de Tang et al., une thrombopénie réfractaire était le premier symptôme, associée à une CIA et un chylothorax5).
Chylothorax : drainage thoracique. Tian et al. ont rapporté une amélioration de l’hypoalbuminémie après microchirurgie pour obstruction de la sortie du canal thoracique6).
Cryptorchidie : orchidopexie à un moment approprié.
QComment traite-t-on les yeux dans le syndrome de Noonan ?
A
La correction de la réfraction par lunettes ou lentilles de contact et le traitement de l’amblyopie sont la base. La chirurgie du strabisme est envisagée en fonction de son degré. L’hypoplasie du nerf optique n’est pas progressive ; il faut éviter un traitement abaissant la pression intraoculaire inapproprié et améliorer la fonction visuelle résiduelle par une correction réfractive adéquate. Le décollement séreux de la rétine associé au colobome de la papille optique peut régresser spontanément et il n’existe pas de traitement standardisé.
La physiopathologie sous-jacente du SN est une signalisation persistante et excessive due à une dérégulation de la voie RAS-MAPK.
PTPN11 (SHP2)
Fonction : Tyrosine phosphatase non réceptrice. Possède un mécanisme d’auto-inhibition par interaction intramoléculaire entre le domaine N-SH2 et le domaine PTP.
Effet des mutations NS : Le mécanisme d’auto-inhibition est levé, l’activité phosphatase est augmentée (gain de fonction). La signalisation RAS-MAPK est hyperactivée 2).
Différence avec NSML : Les mutations NSML (syndrome LEOPARD) sont des formes hypoactives de PTPN11, contrairement au NS.
LZTR1
Fonction : Protéine de l’appareil de Golgi appartenant à la superfamille BTB-Kelch. Favorise la polyubiquitination et la dégradation protéasomique de RAS, régulant négativement la signalisation RAS-MAPK 3).
Mutations AD-NS : Concentrées dans le domaine Kelch (surface de reconnaissance du substrat). Augmentent la signalisation RAS-MAPK (gain de fonction) 1)3).
Mutation AR-NS : due à une mutation de perte de fonction. Elle se lie au complexe RAF1/SHOC2/PP1CB et favorise la phosphorylation de RAF1 Ser259 (inactivation de la voie MAPK) 3).
Les principaux composants de la voie RAS-MAPK sont les suivants :
Régulateurs positifs : SHP2 (PTPN11), SOS1, SOS2. Des mutations gain-de-fonction de ces gènes sont à l’origine du NS.
Facteurs favorisant la signalisation RAS : MRAS, SHOC2, PPP1CB.
Cascade MAPK : BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Régulateurs négatifs : CBL, NF1, LZTR1, SPRED1, SPRED2. Des mutations perte-de-fonction entraînent une hyperactivation de la voie 3).
La voie RAS-MAPK est cruciale pour la différenciation et la prolifération cellulaires ; des mutations gain-de-fonction entraînent une fonction cellulaire anormale dans les tissus de tout le corps, produisant un phénotype complexe impliquant plusieurs organes.
Sur le plan ophtalmologique, l’hypoplasie du nerf optique résulte d’un développement insuffisant des cellules ganglionnaires rétiniennes et des fibres nerveuses. Le colobome de la papille optique est dû à une fermeture incomplète de la fissure optique, avec une prévalence estimée entre 3 et 8 pour 100 000.
En ce qui concerne les tumeurs, les mutations de PTPN11 sont également identifiées comme mutations somatiques dans la JMML (leucémie myélomonocytaire juvénile), le MDS et la LAM ; le risque de tumeur maligne hématologique chez l’enfant est multiplié par 3 dans le syndrome de Noonan 5).
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
Tian et al. (2025) ont rapporté un cas de NS avec mutation LZTR1 c.850C>T présentant une transformation caverneuse de la veine porte, une dysgénésie du canal thoracique, une lymphangiectasie intestinale et une entéropathie exsudative de protéines 6). Une chirurgie microvasculaire pour l’obstruction de la sortie du canal thoracique a été réalisée, permettant la normalisation de l’albumine sérique. Cette observation met en évidence une nouvelle pathologie de la lymphangiogenèse associée à la mutation LZTR1 dans le NS.
Orsolini et al. (2024) ont rapporté un homme de 35 ans porteur de la mutation LZTR1 c.742G>A présentant une FSH élevée et une oligospermie (concentration de spermatozoïdes de 1,5×10⁶/mL) sans antécédent de cryptorchidie7). Ce cas suggère l’existence d’un dysfonctionnement gonadique dû à une atteinte primitive des cellules de Sertoli indépendante de la cryptorchidie, contribuant à la compréhension du mécanisme de l’infertilité masculine dans le syndrome de Noonan.
LZTR1 est également un gène responsable de la schwannomatose, et les recherches sur le chevauchement clinique entre NS, NF1 et la schwannomatose progressent 3). La relation biologique entre RIT1 et LZTR1 attire également l’attention, et il a été suggéré que des mutations de l’un ou l’autre pourraient entraîner une accumulation de RIT1 et contribuer à une hyperactivation de la voie MAPK 3).
Concernant la corrélation génotype-phénotype, son établissement par des cohortes plus larges est un défi futur. L’identification de facteurs prédictifs du pronostic ophtalmologique et le développement de cibles thérapeutiques spécifiques à la NS sont attendus.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.