Das Noonan-Syndrom (NS) ist eine genetische Erkrankung (RASopathie), die durch Genmutationen im RAS-MAPK-Signalweg verursacht wird. Es wurde erstmals 1968 von Jacqueline Noonan systematisch beschrieben.
Die geschätzte Prävalenz liegt bei 1 zu 1.000 bis 2.500 Geburten. Der Erbgang ist in der Regel autosomal-dominant (AD), aber etwa 2/3 der Fälle sind auf Neumutationen (de novo) zurückzuführen. Es sind LZTR1-Mutationen bekannt, die sowohl AD als auch autosomal-rezessiv (AR) vererbt werden können; in einer chinesischen Familie von Zhao et al. zeigte die LZTR1 c.1149+1G>T-Mutation einen AD-Erbgang1).
Charakteristisch sind eine variable Expressivität, bei der der Phänotyp innerhalb derselben Familie mit derselben Mutation stark variiert, und eine unvollständige Penetranz, bei der Träger keinen Phänotyp zeigen. Han & Park (2024) berichteten über einen Fall väterlicher Vererbung der PTPN11 p.Arg498Trp-Mutation, bei dem der Vater asymptomatisch war oder nur eine leichte geistige Behinderung aufwies, und die Rate betroffener Verwandter wird mit 30–40 % angegeben2). De-novo-Mutationen sind mit einem höheren väterlichen Alter assoziiert, und es wird angenommen, dass während der Spermatogenese entstandene Mutationen einen selektiven Vorteil haben.
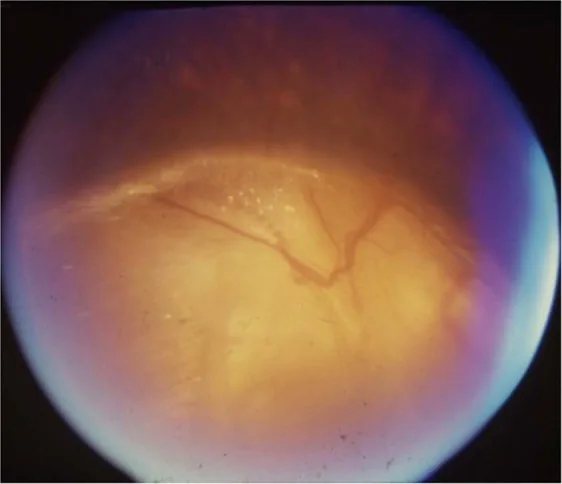
Augenärztlich können verschiedene Anomalien auftreten, darunter Optikushypoplasie, Kolobom der Papille, Refraktionsfehler und Ptosis.
QWie hoch ist die Vererbungswahrscheinlichkeit des Noonan-Syndroms?
A
Der Erbgang ist in der Regel autosomal-dominant, aber etwa 2/3 der Fälle sind auf De-novo-Mutationen (Spontanmutationen) zurückzuführen. Die Vererbung von einem betroffenen Elternteil an ein Kind erfolgt theoretisch mit 50%iger Wahrscheinlichkeit, aber aufgrund unvollständiger Penetranz beträgt die tatsächliche Rate der Symptommanifestation 30–40 %2). Bei positiver Familienanamnese wird eine genetische Beratung empfohlen.
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Charakteristisches Gesicht : breite Stirn, eingesunkene Nasenwurzel, tiefsitzende und nach hinten rotierte Ohren mit dicker Helix. Am stärksten im Säuglings- und Kindesalter ausgeprägt.
Skelett und Hals : Flügelfell, Kiel- oder Trichterbrust, Mamillenhypertelorismus, relative Makrozephalie.
Hämatologisch : verlängerte PT/PTT, Thrombozytenzahl- und Funktionsstörungen, Faktor-XI-Mangel (50%). Eine therapierefraktäre Thrombozytopenie im Neugeborenenalter kann das erste Symptom sein 5).
Genitalien: Kryptorchismus bei Jungen (bis zu 80 %), Unfruchtbarkeit.
Im Fall von Zhao et al. (6,6-jähriges Mädchen) wurden Strabismus, Refraktionsfehler und Nystagmus festgestellt, begleitet von Wachstumshormonmangel 1). Tian et al. (2025) berichteten bei LZTR1-Mutations-NS über kavernöse Transformation der Pfortader, intestinale Lymphangiektasie und proteinverlierende Enteropathie, was die vielfältigen lymphatischen Anomalien zeigt 6).
QWelche Augensymptome treten beim Noonan-Syndrom auf?
A
Es treten vielfältige Befunde auf: Refraktionsfehler (Myopie, Hyperopie, Astigmatismus), Ptosis, Schielen (Strabismus), Nystagmus, Amblyopie. Schwerwiegendere Befunde können Optikushypoplasie, Optikuskolobom und Keratokonus sein. Eine umfassende augenärztliche Untersuchung wird ab der Diagnose empfohlen.
Die Ursache des NS sind Gain-of-Function- (oder Loss-of-Function-)Mutationen in mehreren Genen des RAS-MAPK-Signalwegs. Die Häufigkeit der ursächlichen Gene ist unten angegeben.
Gen
Häufigkeit
Hauptmerkmale
PTPN11
Etwa 50 %
Am häufigsten. Hauptsächlich Gain-of-Function-Mutationen.
SOS1
Etwa 10–13 %
—
RAF1, RIT1
Jeweils etwa 5 %
Berichteter Zusammenhang mit Sehstörungen
LZTR1
etwa 8%
AD- und AR-Vererbung
KRAS, BRAF
jeweils unter 5%
Bericht über niedrigste Sehschärfe bei BRAF-Mutation
LZTR1 ist ein Golgi-Protein der BTB-Kelch-Superfamilie, das die Ubiquitinierung und den Abbau von RAS fördert und so den RAS-MAPK-Signalweg negativ reguliert 3). Die Mutationen bei AD-NS konzentrieren sich auf die Kelch-Domäne (Substraterkennungsfläche) und verstärken den RAS-MAPK-Signalweg 1)3).
Zur Korrelation zwischen Genotyp und ophthalmologischem Phänotyp wurde bei Patienten mit RAF1-, KRAS- und SHOC2-Mutationen eine permanente Sehbehinderung (bester korrigierter binokularer Visus < 0,3) und bei Patienten mit BRAF-Mutation eine minimale Sehschärfe (schlechtes Sehen seit dem Säuglingsalter aufgrund einer Optikushypoplasie) berichtet, jedoch ist eine definitive Korrelation nicht etabliert und die Kohorten sind klein (n=25 bis 105).
Beim NFNS (Neurofibromatose-Noonan-Syndrom) aufgrund einer NF1-Mutation zeigen etwa 25 % der NF1-Patienten Noonan-ähnliche Merkmale, wobei sich die Phänotypen beider Erkrankungen überschneiden 4).
QÄndert sich die Schwere der Augensymptome je nach Genart?
A
Bei Patienten mit RAF1-, KRAS- und SHOC2-Mutationen wurde über dauerhafte Sehbehinderungen berichtet, und bei Patienten mit BRAF-Mutationen wurde eine schlechte Sehkraft ab dem Säuglingsalter aufgrund einer Optikushypoplasie beschrieben. Andererseits gibt es Berichte, dass bei Patienten mit PTPN11-Mutationen keine Sehbehinderung festgestellt wurde. Allerdings sind die Kohorten, die diese Korrelationen zeigen, klein, und derzeit ist keine definitive Genotyp-Phänotyp-Korrelation etabliert.
Die Diagnose des NS wird von einem klinischen Genetiker anhand formaler Diagnosekriterien gestellt, die auf klinischen Befunden wie charakteristischem Gesichtsausdruck, Herzerkrankungen und Kleinwuchs basieren.
Zur Bestätigungsdiagnose wird eine molekulargenetische Untersuchung mittels RAS-MAPK-Signalweg-Genpanel (PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1 usw.) verwendet2)3). Auch die Exomsequenzierung (WES) wird eingesetzt1)4). Es ist zu beachten, dass LZTR1 möglicherweise nicht im NS-Genpanel enthalten ist3).
NFNS (Neurofibromatose-Noonan-Syndrom) : NF1-Mutation führt zu Überlappung der Phänotypen NS+NF1. Bei ≥6 Café-au-lait-Flecken oder Größe ≥5 mm auf NF1-Koinzidenz prüfen 4). Farncombe et al. berichteten über plexiforme Neurofibrome bei NS-Patienten mit LZTR1-Mutation, was eine klinische Überlappung mit NF1 zeigt 3).
Differenzialdiagnose des Optikuskoloboms : peripapilläres Staphylom, Morning-Glory-Syndrom, papilläres PFV/PHPV, Megalopapille.
Andere Syndrome : CHARGE-Syndrom, Turner-Syndrom, kardio-fazio-kutanes Syndrom (CFC), Costello-Syndrom, Legius-Syndrom.
Refraktionskorrektur : Korrektur von Refraktionsfehlern mit Brille oder Kontaktlinsen.
Amblyopiebehandlung : Behandlung der Amblyopie durch Abdecken des gesunden Auges usw.
Schieloperation : Je nach Ausmaß des Schielens wird eine Operation in Betracht gezogen.
Optikushypoplasie: Nicht progredient, Sehschärfe und Gesichtsfeld verändern sich nicht, wenn kein Glaukom hinzukommt. Vorsicht mit unnötiger drucksenkender Therapie. Versuch der Verbesserung der verbleibenden Sehfunktion durch geeignete Refraktionskorrektur.
Seröse Netzhautablösung im Zusammenhang mit einem Optikusdiskuskolobom : Es gibt keine etablierte Behandlung, und in einigen Fällen kommt es zu einer spontanen Rückbildung. Bei einer rhegmatogenen Netzhautablösung wird eine entsprechende Operation in Betracht gezogen.
Kleinwuchs : Wachstumshormontherapie (GH). Bei NS-Patienten kann ein GH-Mangel vorliegen1). Bei NFNS1 besteht jedoch unter GH-Gabe ein Risiko der Vergrößerung von Neurofibromen, sodass eine sorgfältige Abwägung erforderlich ist4).
Herzerkrankungen: Ballondilatation oder chirurgische Korrektur bei Pulmonalklappenstenose. Überwachung und medikamentöse Behandlung der hypertrophen Kardiomyopathie. Hypertrophe Kardiomyopathie bei Kindern unter 2 Jahren birgt das höchste Risiko für Herztod5).
Blutauffälligkeiten : Gerinnungsfaktorsubstitution. Präoperatives Blutungsrisiko-Assessment. Im Neugeborenenfall von Tang et al. war eine refraktäre Thrombozytopenie das erste Symptom, begleitet von ASD und Chylothorax5).
Chylothorax : Thoraxdrainage. Tian et al. berichteten über eine Verbesserung der Hypoalbuminämie nach mikrochirurgischer Behandlung einer Obstruktion des Ductus thoracicus-Ausgangs6).
Kryptorchismus : Orchidopexie zum geeigneten Zeitpunkt.
QWie wird die Augenbehandlung beim Noonan-Syndrom durchgeführt?
A
Die Korrektur von Refraktionsfehlern mit Brille oder Kontaktlinsen und die Behandlung von Amblyopie sind die Grundlage. Schielen wird je nach Ausprägung operativ behandelt. Die Optikushypoplasie ist nicht progredient; eine unnötige drucksenkende Therapie sollte vermieden werden, und die verbleibende Sehfunktion wird durch geeignete Refraktionskorrektur verbessert. Eine seröse Netzhautablösung im Rahmen eines Optikuskoloboms kann sich spontan zurückbilden; es gibt keine standardisierte Behandlung.
Die zugrundeliegende Pathophysiologie des NS ist eine anhaltende und übermäßige Signalübertragung aufgrund einer Fehlregulation des RAS-MAPK-Signalwegs.
PTPN11 (SHP2)
Funktion: Nicht-Rezeptor-Tyrosinphosphatase. Besitzt einen Auto-Inhibitionsmechanismus durch intramolekulare Interaktion zwischen der N-SH2-Domäne und der PTP-Domäne.
Auswirkungen von NS-Mutationen: Der Auto-Inhibitionsmechanismus wird aufgehoben, die Phosphataseaktivität ist erhöht (Funktionsgewinn). Der RAS-MAPK-Signalweg wird überaktiviert 2).
Unterschied zu NSML: NSML-Mutationen (LEOPARD-Syndrom) sind hypomorphe Formen von PTPN11, im Gegensatz zu NS.
LZTR1
Funktion: Golgi-Protein der BTB-Kelch-Superfamilie. Fördert die Polyubiquitinierung und den proteasomalen Abbau von RAS und reguliert den RAS-MAPK-Signalweg negativ 3).
AD-NS-Mutationen: Konzentriert in der Kelch-Domäne (Substraterkennungsfläche). Steigern den RAS-MAPK-Signalweg (Funktionsgewinn) 1)3).
AR-NS-Mutation : durch Funktionsverlustmutation. Sie bindet an den RAF1/SHOC2/PP1CB-Komplex und fördert die Phosphorylierung von RAF1 Ser259 (MAPK-Signal-Inaktivierung) 3).
Die Hauptkomponenten des RAS-MAPK-Signalwegs sind:
Positive Regulatoren : SHP2 (PTPN11), SOS1, SOS2. Gain-of-Function-Mutationen dieser Gene verursachen NS.
RAS-Signalverstärker : MRAS, SHOC2, PPP1CB.
MAPK-Kaskade : BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Negative Regulatoren : CBL, NF1, LZTR1, SPRED1, SPRED2. Funktionsverlustmutationen führen zu einer Signalverstärkung 3).
Der RAS-MAPK-Signalweg ist für Zelldifferenzierung und -proliferation essenziell; Gain-of-Function-Mutationen führen zu abnormaler Zellfunktion in allen Körpergeweben und verursachen einen komplexen Phänotyp mit Beteiligung mehrerer Organe.
Zu den ophthalmologischen Befunden gehört die Optikushypoplasie, die auf eine unzureichende Entwicklung retinaler Ganglienzellen und Nervenfasern zurückzuführen ist. Das Kolobom der Papille entsteht durch einen unvollständigen Verschluss der Augenbecherspalte, mit einer Prävalenz von 3–8/100.000.
Im Zusammenhang mit Tumoren werden PTPN11-Mutationen auch als somatische Mutationen bei JMML (juveniler myelomonozytärer Leukämie), MDS und AML identifiziert; das Risiko für hämatologische Malignome im Kindesalter ist beim Noonan-Syndrom um das Dreifache erhöht 5).
7. Aktuelle Forschung und Zukunftsperspektiven (Berichte aus der Forschungsphase)
Tian et al. (2025) berichteten über einen Fall von NS mit LZTR1 c.850C>T-Mutation, der eine kavernöse Transformation der Pfortader, eine Dysplasie des Ductus thoracicus, intestinale Lymphangiektasie und eine proteinverlierende Enteropathie aufwies 6). Eine mikrochirurgische Behandlung der Ductus-thoracicus-Ausgangsobstruktion wurde durchgeführt, wodurch eine Normalisierung des Serumalbumins erreicht wurde. Dies zeigt eine neue Pathologie der Lymphangiogenese im Zusammenhang mit der LZTR1-Mutation bei NS.
Orsolini et al. (2024) berichteten über einen 35-jährigen Mann mit LZTR1 c.742G>A-Mutation, der ohne Kryptorchismus in der Vorgeschichte erhöhtes FSH und Oligospermie (Spermienkonzentration 1,5×10⁶/mL) aufwies7). Dieser Fall deutet auf das Vorliegen einer von Kryptorchismus unabhängigen gonadalen Dysfunktion aufgrund einer primären Sertoli-Zell-Störung hin und trägt zur Aufklärung des Mechanismus der männlichen Infertilität beim Noonan-Syndrom bei.
Genotyp-Phänotyp-Korrelation und zukünftige Herausforderungen
LZTR1 ist auch ein ursächliches Gen für Schwannomatose, und die Forschung zur klinischen Überlappung von NS, NF1 und Schwannomatose schreitet voran 3). Auch die biologische Beziehung zwischen RIT1 und LZTR1 wird beachtet, und es wird vermutet, dass Mutationen in einem von beiden zu einer Akkumulation von RIT1 und einer Überaktivierung des MAPK-Signalwegs beitragen könnten 3).
Hinsichtlich der Genotyp-Phänotyp-Korrelation ist deren Etablierung durch größere Kohorten eine zukünftige Herausforderung. Die Identifizierung von Prädiktoren für die ophthalmologische Prognose und die Entwicklung NS-spezifischer Therapieziele werden erwartet.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.