Noonan sendromu (NS), RAS-MAPK sinyal yolundaki gen mutasyonlarından kaynaklanan kalıtsal bir hastalıktır (RASopati). İlk kez 1968 yılında Jacqueline Noonan tarafından sistematik olarak tanımlanmıştır.
Tahmini prevalansın 1.000 ila 2.500 doğumda 1 olduğu düşünülmektedir. Kalıtım şekli genellikle otozomal dominanttır (AD), ancak vakaların yaklaşık 2/3’ü yeni (de novo) mutasyonlardan kaynaklanır. LZTR1 mutasyonlarının hem AD hem de AR (otozomal resesif) formları aldığı bilinmektedir; Zhao ve ark.‘nın Çin ailesinde LZTR1 c.1149+1G>T mutasyonu AD kalıtım göstermiştir1).
Aynı mutasyona sahip ailelerde fenotipin büyük ölçüde farklılık gösterdiği “değişken ekspresivite” ve taşıyıcıların fenotip göstermediği “eksik penetrans” karakteristiktir. Han & Park (2024), PTPN11 p.Arg498Trp mutasyonuna sahip babadan kalıtılan bir vaka bildirmiştir; baba asemptomatik veya sadece hafif zihinsel engelli olup, etkilenen akrabaların oranı %30-40 olarak belirtilmiştir2). De novo mutasyonlar babanın ileri yaşı ile ilişkilidir ve spermatogenez sırasında ortaya çıkan mutasyonların seçici avantaja sahip olduğu düşünülmektedir.
Genellikle otozomal dominant kalıtım gösterir, ancak yaklaşık 2/3’ü de novo mutasyon (kendiliğinden oluşan) kaynaklıdır. Etkilenen bir ebeveynden çocuğa geçiş teorik olarak %50 olasılıkla olur, ancak eksik penetrans nedeniyle semptomların gerçekte ortaya çıkma oranı %30-40 olarak kabul edilir2). Aile öyküsü varsa genetik danışmanlık önerilir.
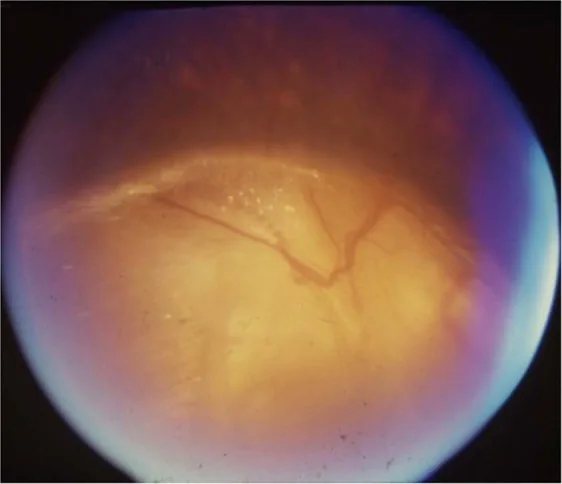
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Dış göz bulguları: Pitozis (göz kapağı düşüklüğü), hipertelorizm (gözler arası mesafenin artması), aşağı eğimli palpebral fissürler (dış göz kapağı düşüklüğü), yüksek kemerli kaşlar, epikantus (iç göz kapağı kıvrımı).
Ön segment bulguları: Keratokonus, belirgin kornea sinirleri, mavi/yeşil-mavi iris, posterior embriyotokson, katarakt.
Nöro-oftalmolojik bulgular: Şaşılık, nistagmus, ambliyopi, optik sinir hipoplazisi, çukurlaşma, koloboma dahil bilateral optik disk anormallikleri.
Sistemik bulgular
Karakteristik yüz görünümü: Geniş alın, burun kökünde çöküklük, kalın kulak kepçesi ile birlikte aşağıda ve arkaya dönük kulaklar. Bebeklik ve çocukluk döneminde en belirgindir.
İskelet ve boyun: Kanat boyun, güvercin göğsü/huni göğüs, meme başı aralığının açık olması, görece büyük kafa.
Hematolojik: PT/PTT uzaması, trombosit sayısı ve fonksiyon bozukluğu, faktör XI eksikliği (%50). Yenidoğan döneminde tedaviye dirençli trombositopeni ilk belirti olabilir 5).
Zhao ve ark.‘nın vakasında (6,6 yaşında kız çocuğu) şaşılık, kırma kusuru ve nistagmus gözlenmiş ve büyüme hormonu eksikliği eşlik etmiştir1). Tian ve ark. (2025), LZTR1 mutasyonlu Noonan sendromunda portal ven kavernöz dönüşümü, intestinal lenfanjiektazi ve protein kaybettiren enteropati bildirmiş olup lenfatik malformasyonların çeşitli patolojilerini ortaya koymuştur6).
QNoonan sendromunda gözün hangi belirtileri görülür?
NS’nin nedeni, RAS-MAPK yolunda yer alan birden fazla gendeki fonksiyon kazandıran (veya kaybettiren) mutasyonlardır. Nedensel genlerin sıklığı aşağıda gösterilmiştir.
Gen
Sıklık
Başlıca özellikler
PTPN11
Yaklaşık %50
En sık. Başlıca işlev kazandıran mutasyonlar
SOS1
Yaklaşık %10-13
—
RAF1, RIT1
Her biri yaklaşık %5
Görme bozukluğu ile ilişkili olduğu bildirilmiştir
LZTR1
Yaklaşık %8
AD ve AR kalıtım şekilleri
KRAS, BRAF
Her biri %5’ten az
BRAF mutasyonunda en düşük görme keskinliği bildirilmiştir
LZTR1, BTB-Kelch süper ailesine ait bir Golgi proteini olup, RAS’ın ubikitinasyonunu ve parçalanmasını teşvik ederek RAS-MAPK sinyalini negatif yönde düzenler 3). AD-NS mutasyonları Kelch alanında (substrat tanıma yüzeyi) yoğunlaşır ve RAS-MAPK sinyalini artırır 1)3).
Genotip ve oftalmolojik fenotip arasındaki ilişkiye dair, RAF1, KRAS ve SHOC2 mutasyonu olan hastalarda kalıcı görme bozukluğu (iki gözde en iyi düzeltilmiş görme keskinliği < 0,3) ve BRAF mutasyonu olan hastalarda en düşük görme keskinliği (optik sinir hipoplazisine bağlı bebeklikten itibaren kötü görme) bildirilmiştir, ancak kesin bir ilişki kurulamamıştır ve kohortlar küçüktür (n=25-105).
NF1 mutasyonuna bağlı NFNS’de (nörofibromatozis-Noonan sendromu), NF1 hastalarının yaklaşık %25’i Noonan benzeri özellikler gösterir ve iki hastalığın fenotipi örtüşür 4).
QGöz semptomlarının şiddeti genin türüne göre değişir mi?
A
RAF1, KRAS ve SHOC2 mutasyonu olan hastalarda kalıcı görme bozukluğu bildirilmiş olup, BRAF mutasyonu olan hastalarda optik sinir hipoplazisine bağlı bebeklik döneminden itibaren kötü görme rapor edilmiştir. Öte yandan, PTPN11 mutasyonu olan hastalarda görme bozukluğu gözlenmediğini belirten çalışmalar da vardır. Ancak, bu korelasyonları gösteren kohortlar küçük ölçeklidir ve şu anda kesin bir genotip-fenotip ilişkisi kurulmamıştır.
NS tanısı, karakteristik yüz görünümü, kalp hastalığı, kısa boy gibi klinik bulgulara dayanan resmi tanı kriterlerine göre klinik genetik uzmanı tarafından değerlendirilir.
Kesin tanı için RAS-MAPK yolu ile ilişkili gen paneli (PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1 vb.) kullanılarak moleküler genetik test yapılır2)3). Tüm ekzom dizileme (WES) de kullanılır1)4). NS gen panelinde LZTR1’in bulunmama ihtimaline karşı dikkatli olunmalıdır3).
NFNS (Nörofibromatozis-Noonan sendromu) : NF1 mutasyonu nedeniyle NS+NF1 fenotipleri örtüşür. Altı veya daha fazla café-au-lait lekesi veya 5 mm’den büyük lezyonlar varlığında NF1 birlikteliği araştırılır4). Farncombe ve ark., LZTR1 mutasyonu olan NS hastalarında pleksiform nörofibrom saptamış ve NF1 ile klinik örtüşme bildirmiştir3).
Optik disk kolobomunun ayırıcı tanısı: Peripapiller stafilom, sabah zaferi sendromu, papiller PFV/PHPV, megapapilla.
Refraksiyon düzeltmesi: Kırma kusurlarının gözlük veya kontakt lens ile düzeltilmesi.
Ambliyopi tedavisi: Sağlıklı gözün kapatılması gibi yöntemlerle ambliyopi tedavisi.
Şaşılık cerrahisi: Şaşılığın derecesine göre cerrahi düşünülür.
Optik sinir hipoplazisi: İlerleyici değildir, glokom eşlik etmezse görme keskinliği ve alanı değişmez. Gereksiz göz içi basıncı düşürücü tedaviden kaçının. Uygun refraksiyon düzeltmesi ile kalan görme fonksiyonunu iyileştirmeye çalışın.
Optik disk kolobomuna eşlik eden seröz retina dekolmanı: Belirlenmiş bir tedavisi yoktur ve kendiliğinden gerileyen vakalar da vardır. Regmatojen retina dekolmanında buna uygun cerrahi tedavi düşünülür.
Kalp hastalıkları: Pulmoner kapak darlığı için balon dilatasyonu/cerrahi onarım. Hipertrofik kardiyomiyopatinin takibi/ilaç tedavisi. 2 yaş altı hipertrofik kardiyomiyopati, kardiyak ölüm için en yüksek riski oluşturur5).
Kan anormallikleri: Pıhtılaşma faktörü takviyesi. Ameliyat öncesi kanama riski değerlendirmesi. Tang ve ark.‘nın yenidoğan vakasında, ilk belirti dirençli trombositopeniydi ve hastada ASD ile şilotoraks birlikte görülüyordu5).
Şilotoraks: Plevral drenaj. Tian ve ark., torasik kanal çıkış tıkanıklığına yönelik mikroskobik cerrahi ile hipoalbümineminin düzeldiğini bildirmiştir6).
İnmemiş testis: Uygun zamanda testis fiksasyonu (orşiopeksi).
QNoonan sendromunda göz tedavisi nasıl yapılır?
A
Gözlük ve kontakt lenslerle refraksiyon düzeltmesi ve ambliyopi tedavisi temeldir. Şaşılık durumunda derecesine göre cerrahi düşünülür. Optik sinir hipoplazisi ilerleyici değildir; gereksiz göz içi basıncı düşürücü tedaviden kaçınılmalı ve uygun refraksiyon düzeltmesi ile kalan görme fonksiyonu iyileştirilmeye çalışılmalıdır. Optik disk kolobomuna bağlı seröz retina dekolmanı kendiliğinden gerileyebilir ve kesin bir tedavisi yoktur.
NS’nin temel patofizyolojisi, RAS-MAPK yolunun düzensizliği nedeniyle sürekli ve aşırı sinyal iletimidir.
PTPN11 (SHP2)
Fonksiyon: Reseptör olmayan tirozin fosfataz. N-SH2 alanı ve PTP alanı arasındaki molekül içi etkileşim yoluyla kendi kendini inhibe etme mekanizmasına sahiptir.
NS mutasyonunun etkileri: Kendi kendini inhibe etme mekanizması ortadan kalkar ve fosfataz aktivitesi artar (işlev kazanımı). RAS-MAPK sinyali aşırı aktif hale gelir 2).
Fonksiyon: BTB-Kelch süper ailesine ait Golgi proteini. RAS’ın poliubikitinasyonunu ve proteazom yıkımını teşvik ederek RAS-MAPK sinyalini negatif yönde düzenler 3).
AR-NS mutasyonu: Fonksiyon kaybı tipi mutasyon nedeniyle oluşur. Ayrıca RAF1/SHOC2/PP1CB kompleksine bağlanarak RAF1 Ser259 fosforilasyonunu teşvik eden (MAPK sinyalini inaktive eden) bir işleve sahiptir3).
RAS-MAPK yolunun ana bileşenleri aşağıdaki gibidir.
Pozitif düzenleyiciler: SHP2 (PTPN11), SOS1, SOS2. Bu genlerdeki işlev kazandıran mutasyonlar NS’ye neden olur.
Negatif düzenleyiciler: CBL, NF1, LZTR1, SPRED1, SPRED2. Fonksiyon kaybı mutasyonları sinyal artışına neden olur3).
RAS-MAPK yolu, hücre farklılaşması ve çoğalması için son derece önemlidir ve işlev kazandıran mutasyonlar, tüm vücut dokularında anormal hücresel işlevlere yol açarak çoklu organları etkileyen karmaşık bir fenotipe neden olur.
Oftalmolojik durum olarak, optik sinir hipoplazisi, retina ganglion hücreleri ve sinir liflerinin gelişimsel yetersizliğinden kaynaklanır. Optik disk kolobomu, optik fissürün kapanma kusuruna bağlıdır ve prevalansı 3-8/100.000 olarak bildirilmiştir.
Tümörlerle ilişkili olarak, PTPN11 mutasyonu JMML (juvenil miyelomonositik lösemi), MDS ve AML’de somatik mutasyon olarak da tanımlanmıştır ve NS’de çocukluk çağı hematolojik malignite riskinin 3 kat olduğu bildirilmiştir 5).
7. En son araştırmalar ve geleceğe yönelik bakış (araştırma aşamasındaki raporlar)
Tian ve ark. (2025), LZTR1 c.850C>T mutasyonu NS’li bir olguda portal venöz kavernöz transformasyon, torasik kanal hipoplazisi, intestinal lenfanjiektazi ve protein kaybettiren enteropati bildirmiştir6). Torasik kanal çıkış obstrüksiyonu için mikrocerrahi uygulanmış ve serum albümin düzeylerinde normalleşme sağlanmıştır. Bu, LZTR1 mutasyonu NS’ye eşlik eden lenfatik malformasyonun yeni bir patofizyolojisini gösteren bir bulgudur.
Orsolini ve ark. (2024), LZTR1 c.742G>A mutasyonuna sahip 35 yaşında bir erkekte, inmemiş testis öyküsü olmaksızın yüksek FSH ve oligospermi (sperm konsantrasyonu 1.5×10⁶/mL) bildirmiştir7). Bu olgu, inmemiş testisten bağımsız olarak primer Sertoli hücre hasarına bağlı gonadal disfonksiyonun varlığını düşündürmekte olup, NS’de erkek infertilitesinin mekanizmasının aydınlatılmasına katkı sağlayan bir bulgudur.
Genotip-fenotip korelasyonu ve gelecekteki zorluklar
LZTR1 aynı zamanda schwannomatozisin (schwannomatosis) neden olan genidir ve NS, NF1 ve schwannomatozis arasındaki klinik örtüşme üzerine araştırmalar devam etmektedir3). RIT1 ve LZTR1 arasındaki biyolojik ilişki de dikkat çekmektedir; her iki gendeki mutasyonun RIT1 birikimine yol açarak MAPK sinyalinin aşırı aktivasyonuna katkıda bulunabileceği öne sürülmüştür3).
Genotip-fenotip korelasyonu için daha büyük kohortlarla doğrulama gelecekteki bir zorluktur. Oftalmolojik prognoz belirleyicilerinin tanımlanması ve NS’ye özgü tedavi hedeflerinin geliştirilmesi beklenmektedir.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.