กลุ่มอาการนูนันเป็น RASopathy เกิดจากการกลายพันธุ์ของยีนในวิถี RAS-MAPK พบได้ 1 ใน 1,000–2,500 คนเกิดมีชีพ
ลักษณะสำคัญสามประการ ได้แก่ ใบหน้าที่มีลักษณะเฉพาะ ตัวเตี้ย และโรคหัวใจพิการแต่กำเนิด ร่วมกับอาการอื่นๆ ที่หลากหลายในหลายอวัยวะ
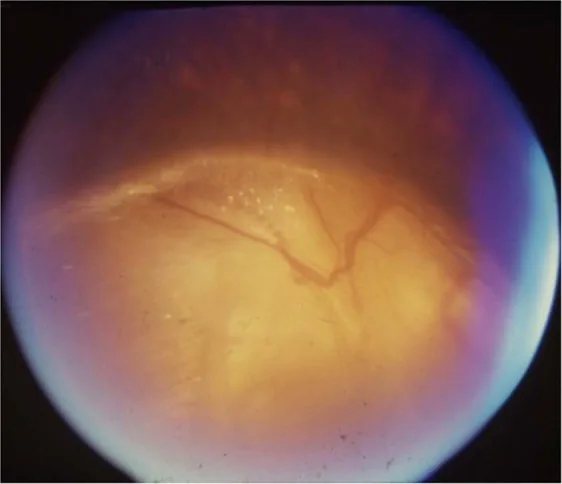
ผลตรวจทางจักษุวิทยา ได้แก่ ภาวะผิดปกติของการหักเหแสง ตาเหล่ ตากระตุก หนังตาตก เส้นประสาทตา พัฒนาน้อย และคอโลโบมาของจานประสาทตา
มียีนก่อโรคหลายชนิด โดย PTPN11 คิดเป็นประมาณ 50% การกลายพันธุ์ของ LZTR1 สามารถเป็นได้ทั้งแบบเด่นและด้อยบนออโตโซม
แนะนำให้ประเมินทางจักษุวิทยาอย่างครอบคลุมเมื่อวินิจฉัย และติดตามผลเป็นประจำทุกปีหลังจากนั้น
ภาวะเส้นประสาทตา พัฒนาน้อยไม่มีการดำเนินโรค และการแก้ไขค่าสายตาที่เหมาะสมร่วมกับการรักษาภาวะตาขี้เกียจ เป็นพื้นฐานในการปรับปรุงการมองเห็น
ไม่มีการรักษาให้หายขาด และจำเป็นต้องมีการจัดการแบบสหสาขาวิชาชีพตลอดชีวิต
กลุ่มอาการนูนัน (Noonan syndrome; NS) เป็นโรคทางพันธุกรรม (RASopathy) ที่เกิดจากการกลายพันธุ์ของยีนในวิถีสัญญาณ RAS-MAPK รายงานอย่างเป็นระบบครั้งแรกโดย Jacqueline Noonan ในปี ค.ศ. 1968
ประมาณการความชุกอยู่ที่ 1 ใน 1,000 ถึง 2,500 คนเกิด รูปแบบการถ่ายทอดทางพันธุกรรมโดยทั่วไปเป็นแบบออโตโซมอลโดมิแนนต์ (AD) แต่ประมาณ 2 ใน 3 ของผู้ป่วยเกิดจากการกลายพันธุ์ใหม่ (de novo) การกลายพันธุ์ของยีน LZTR1 ที่เป็นได้ทั้งแบบ AD และ AR (ออโตโซมอลรีเซสซีฟ) เป็นที่ทราบกันดี ในครอบครัวชาวจีนที่ศึกษาโดย Zhao และคณะ การกลายพันธุ์ LZTR1 c.1149+1G>T แสดงการถ่ายทอดแบบ AD 1) .
กลุ่มอาการนูนันมีลักษณะเฉพาะคือ “การแสดงออกที่แปรผัน” ซึ่งฟีโนไทป์แตกต่างกันอย่างมากภายในครอบครัวที่มีการกลายพันธุ์เดียวกัน และ “การแทรกซึมไม่สมบูรณ์” ซึ่งพาหะไม่แสดงฟีโนไทป์ Han & Park (2024) รายงานกรณีการถ่ายทอดทางบิดาด้วยการกลายพันธุ์ PTPN11 p.Arg498Trp โดยบิดาไม่มีอาการหรือมีความบกพร่องทางสติปัญญาเพียงเล็กน้อย และญาติที่ได้รับผลกระทบ 30-40% 2) การกลายพันธุ์แบบ de novo สัมพันธ์กับอายุบิดาที่มากขึ้น และเชื่อว่าการกลายพันธุ์ที่เกิดขึ้นระหว่างการสร้างอสุจิมีข้อได้เปรียบในการคัดเลือก
ทางจักษุวิทยา กลุ่มอาการนูนันอาจมีความผิดปกติหลายอย่างร่วมด้วย เช่น ภาวะเส้นประสาทตาเจริญไม่เต็มที่ คอโลโบมาของจานประสาทตา ความผิดปกติของการหักเหของแสง และหนังตาตก
Q
กลุ่มอาการนูนันถ่ายทอดทางพันธุกรรมมากแค่ไหน?
A
โดยปกติแล้วจะแสดงการถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่น แต่ประมาณ 2 ใน 3 ของผู้ป่วยเกิดจากการกลายพันธุ์ใหม่ (ที่เกิดขึ้นเอง) ในทางทฤษฎี ความน่าจะเป็นในการถ่ายทอดจากพ่อแม่ที่ป่วยไปยังบุตรคือ 50% แต่เนื่องจากการแทรกซึมไม่สมบูรณ์ สัดส่วนที่แท้จริงของการแสดงอาการจึงอยู่ที่ 30-40% 2) หากมีประวัติครอบครัว แนะนำให้ปรึกษาทางพันธุกรรม
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
ภาพแสดงลักษณะใบหน้าที่เกี่ยวข้องกับกลุ่มอาการนูนัน
การมองเห็น ลดลงการมองเห็น อาจมีตั้งแต่ทารกเนื่องจากภาวะเส้นประสาทตา พัฒนาน้อยหรือคอโลโบมาตาเหล่ ความสูงเตี้ย : ความยาวแรกเกิดปกติ แต่ความสูงเตี้ยจะชัดเจนเมื่อโตขึ้นพัฒนาการล่าช้า : ความล่าช้าทางภาษาและความบกพร่องทางสติปัญญาเล็กน้อยอาจเป็นอาการเริ่มแรก
อาการแสดงทางจักษุวิทยา
ความผิดปกติของการหักเหแสง : สายตาสั้น สายตายาว (รวมถึงระดับรุนแรง) และสายตาเอียง พบในผู้ป่วยเกือบทุกราย
อาการแสดงภายนอกลูกตา : หนังตาตก ระยะห่างระหว่างเบ้าตา กว้าง (hypertelorism) รอยแยกเปลือกตาลาดลง (down-slanting palpebral fissures) คิ้วโค้งสูง และรอยพับหนังตาชั้นใน (epicanthus)
อาการแสดงส่วนหน้าของลูกตา : กระจกตา รูปกรวย เส้นประสาทกระจกตา เด่นชัด ม่านตา สีฟ้าหรือฟ้าเขียว วงแหวนตัวอ่อนด้านหลัง (posterior embryotoxon) และต้อกระจก
ผลตรวจทางประสาทจักษุวิทยา : ความผิดปกติของจานประสาทตา ทั้งสองข้าง ได้แก่ ตาเหล่ อาตา ตามัว เส้นประสาทตา พัฒนาน้อยกว่าปกติ เส้นประสาทตา บุ๋ม และคอโลโบมา
อาการแสดงทั่วร่างกาย
ลักษณะใบหน้าที่เด่นชัด : หน้าผากกว้าง สันจมูกบุ๋ม หูต่ำและหมุนไปด้านหลังพร้อมกับขอบหูที่หนา เด่นชัดที่สุดในวัยทารกและวัยเด็ก
ระบบหัวใจและหลอดเลือด : ลิ้นหัวใจพัลโมนารีตีบ (50-60%), กล้ามเนื้อหัวใจหนาผิดปกติ (20%), รูรั่วที่ผนังหัวใจห้องบน (6-10%), รูรั่วที่ผนังหัวใจห้องล่าง, การสร้างท่อน้ำเหลืองไม่สมบูรณ์.
โครงกระดูกและคอ : คอเป็นปีก, หน้าอกนกพิราบหรือหน้าอกกรวย, หัวนมแยกห่าง, ศีรษะโตสัมพัทธ์.
ทางโลหิตวิทยา : PT/PTT ยาวขึ้น ความผิดปกติของจำนวนและการทำงานของเกล็ดเลือด การขาดปัจจัย XI (50%) ภาวะเกล็ดเลือดต่ำที่ดื้อต่อการรักษาในทารกแรกเกิดอาจเป็นอาการเริ่มแรก 5) .
อวัยวะสืบพันธุ์ : อัณฑะไม่ลงถุงในเพศชาย (สูงถึง 80%), ภาวะมีบุตรยาก
กรณีของ Zhao et al. (เด็กหญิงอายุ 6.6 ปี) พบตาเหล่ ภาวะผิดปกติของการหักเหแสง และอาตา ร่วมกับภาวะขาดฮอร์โมนการเจริญเติบโต 1) Tian et al. (2025) รายงานการเปลี่ยนแปลงเป็นโพรงเลือดดำพอร์ทัล การขยายตัวของท่อน้ำเหลืองในลำไส้ และภาวะลำไส้สูญเสียโปรตีนใน NS ที่มีการกลายพันธุ์ LZTR1 แสดงให้เห็นความหลากหลายของความผิดปกติของท่อน้ำเหลือง 6)
Q
อาการทางตาของกลุ่มอาการนูนันมีอะไรบ้าง?
A
พบอาการที่หลากหลาย เช่น ภาวะผิดปกติของการหักเหแสง (สายตาสั้น สายตายาว สายตาเอียง ) หนังตาตก การเบี่ยงเบนของตา (ตาเหล่ ) อาตา และตามัว อาการที่รุนแรงมากขึ้นอาจรวมถึงภาวะเส้นประสาทตา พัฒนาน้อยกว่าปกติ คอโลโบมาของจานประสาทตา และภาวะกระจกตา รูปกรวย แนะนำให้ประเมินตาอย่างครอบคลุมตั้งแต่การวินิจฉัย
สาเหตุของ NS คือการกลายพันธุ์แบบ gain-of-function (หรือ loss-of-function) ในยีนหลายตัวที่เกี่ยวข้องกับวิถี RAS-MAPK ความถี่ของยีนก่อโรคแสดงไว้ด้านล่าง
ยีน ความถี่ ลักษณะสำคัญ PTPN11 ประมาณ 50% พบบ่อยที่สุด การกลายพันธุ์แบบ gain-of-function เป็นหลัก SOS1 ประมาณ 10-13% — RAF1, RIT1 ประมาณ 5% ต่อยีน มีรายงานความสัมพันธ์กับความบกพร่องทางการมองเห็น LZTR1 ประมาณ 8% รูปแบบการถ่ายทอดทางพันธุกรรมแบบ AD และ AR KRAS, BRAF แต่ละชนิดน้อยกว่า 5% การกลายพันธุ์ของ BRAF รายงานว่าสัมพันธ์กับสายตาที่แย่ที่สุด
LZTR1 เป็นโปรตีนของกอลจิที่อยู่ในซูเปอร์แฟมิลี BTB-Kelch ซึ่งส่งเสริมการยูบิควิติเนชันและการย่อยสลายของ RAS เพื่อควบคุมสัญญาณ RAS-MAPK ในทางลบ 3) การกลายพันธุ์แบบ AD-NS จะกระจุกตัวอยู่ในโดเมน Kelch (พื้นผิวที่จดจำซับสเตรต) และเพิ่มสัญญาณ RAS-MAPK 1) 3)
เกี่ยวกับความสัมพันธ์ระหว่างจีโนไทป์และฟีโนไทป์ทางจักษุวิทยา มีรายงานความบกพร่องทางการมองเห็น ถาวร (ค่าสายตาที่ดีที่สุดหลังแก้ไข < 0.3) ในผู้ป่วยที่มีการกลายพันธุ์ RAF1, KRAS และ SHOC2 และค่าสายตาต่ำที่สุด (การมองเห็น ไม่ดีตั้งแต่ทารกเนื่องจากภาวะเส้นประสาทตา พัฒนาน้อย) ในผู้ป่วยที่มีการกลายพันธุ์ BRAF แต่ยังไม่มีการสร้างความสัมพันธ์ที่แน่ชัด และกลุ่มตัวอย่างมีขนาดเล็ก (n=25-105)
ใน NFNS (กลุ่มอาการนิวโรไฟโบรมาทอซิส-นูนัน) เนื่องจากการกลายพันธุ์ NF1 ผู้ป่วย NF1 ประมาณ 25% แสดงลักษณะคล้ายนูนัน และฟีโนไทป์ของทั้งสองโรคทับซ้อนกัน 4)
กลุ่มอาการนูนันเป็นโรคทางพันธุกรรม การป้องกันการเกิดโรคทำได้ยาก หากมีประวัติครอบครัว แนะนำให้รับคำปรึกษาทางพันธุกรรม อาจมีการตรวจก่อนคลอดและการตรวจทางพันธุกรรมก่อนการฝังตัวของตัวอ่อน
Q
ความรุนแรงของอาการทางตาขึ้นอยู่กับชนิดของยีนหรือไม่?
A
มีรายงานความบกพร่องทางการมองเห็น ถาวรในผู้ป่วยที่มีการกลายพันธุ์ RAF1, KRAS และ SHOC2 และความบกพร่องทางการมองเห็น ตั้งแต่ทารกเนื่องจากภาวะเส้นประสาทตา เจริญผิดปกติในผู้ป่วยที่มีการกลายพันธุ์ BRAF ในทางกลับกัน มีรายงานว่าไม่พบความบกพร่องทางการมองเห็น ในผู้ป่วยที่มีการกลายพันธุ์ PTPN11 อย่างไรก็ตาม กลุ่มตัวอย่างที่แสดงความสัมพันธ์เหล่านี้มีขนาดเล็ก และในปัจจุบันยังไม่มีการสร้างความสัมพันธ์ระหว่างจีโนไทป์-ฟีโนไทป์ที่แน่ชัด
การวินิจฉัย NS ดำเนินการตามเกณฑ์การวินิจฉัยอย่างเป็นทางการโดยอาศัยผลการตรวจทางคลินิก เช่น ใบหน้าที่มีลักษณะเฉพาะ โรคหัวใจ และความสูงเตี้ย และประเมินโดยผู้เชี่ยวชาญด้านพันธุศาสตร์คลินิก
เพื่อการวินิจฉัยที่แน่ชัด ใช้การตรวจทางพันธุศาสตร์ระดับโมเลกุลด้วยชุดยีนที่เกี่ยวข้องกับวิถี RAS-MAPK (เช่น PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1)2) 3) นอกจากนี้ยังใช้การหาลำดับเอ็กโซมทั้งหมด (WES)1) 4) ควรสังเกตว่าชุดยีน NS อาจไม่รวม LZTR13)
แนะนำให้ประเมินทางจักษุวิทยาอย่างครอบคลุมเมื่อวินิจฉัย หลังจากนั้นติดตามผลปีละครั้ง
การตรวจวัดสายตา สายตาเอียง หรือไม่การตรวจตาเหล่ : ประเมินตำแหน่งตาและการเคลื่อนไหวของลูกตาการตรวจอวัยวะภายในลูกตา : ประเมินลักษณะของจานประสาทตา
อัตราส่วน DM/DD (ระยะห่างระหว่างจานประสาทตา กับรอยบุ๋มจอตา /เส้นผ่านศูนย์กลางจานประสาทตา ): 3 ขึ้นไปสงสัยภาวะเส้นประสาทตา พัฒนาน้อย 4 ขึ้นไปมีโอกาสสูงเครื่องหมายวงแหวนคู่ : วงแหวนเม็ดสีรอบหัวประสาทตาการตรวจด้วยเครื่องเอกซเรย์การเชื่อมประสานด้วยแสง (OCT ) : การวัดความหนาของชั้นเส้นใยประสาทจอประสาทตา รอบหัวประสาทตา (cpRNFL) มีประโยชน์การตรวจคลื่นแม่เหล็กไฟฟ้าสมอง (MRI) : การประเมินความผิดปกติของระบบประสาทส่วนกลาง ประมาณ 15% มีความผิดปกติของ infundibulum ต่อมใต้สมอง
ผลการตรวจด้วยกล้องตรวจตา : ความบกพร่องของหัวประสาทตาและคอรอยด์ -จอประสาทตา ที่อยู่ด้านล่างเป็นหลัก ความผิดปกติของแนวเส้นเลือดการตรวจภาพ : การวินิจฉัยที่แน่นอนด้วยอัลตราซาวนด์, MRI, CT และ OCT ค้นหาความผิดปกติของกะโหลกศีรษะร่วมด้วยโดย MRI/CT สมอง
NFNS (Neurofibromatosis-Noonan syndrome) : การกลายพันธุ์ของ NF1 ทำให้เกิดฟีโนไทป์ที่ซ้อนทับกันของ NS+NF1 ค้นหาความเกี่ยวข้องของ NF1 หากมีรอยด่าง café-au-lait ≥6 จุด หรือขนาด ≥5 มม.4) Farncombe และคณะรายงาน neurofibroma แบบ plexiform ในผู้ป่วย NS ที่มีการกลายพันธุ์ LZTR1 ซึ่งบ่งชี้ถึงการซ้อนทับทางคลินิกกับ NF1 3) .การวินิจฉัยแยกโรค coloboma ของจานประสาทตา : Staphyloma รอบหัวประสาทตา, กลุ่มอาการ morning glory, PFV /PHPV ที่หัวประสาทตา, macropapillaกลุ่มอาการอื่นๆ : CHARGE syndrome , Turner syndrome, cardiofaciocutaneous syndrome (CFC), Costello syndrome, Legius syndrome
Echocardiogram : การตรวจคัดกรองความผิดปกติของหัวใจการตรวจเลือด : การทดสอบการทำงานของการแข็งตัวของเลือด (PT/APTT, กิจกรรมของปัจจัย XI เป็นต้น) 5) .
NS ไม่มีการรักษาให้หายขาด และการจัดการแบบสหสาขาวิชาชีพโดยมีนักพันธุศาสตร์คลินิกเป็นผู้นำเป็นพื้นฐานของการดูแล
การแก้ไขค่าสายตา : การแก้ไขความผิดปกติของการหักเหของแสง ด้วยแว่นตาหรือคอนแทคเลนส์การรักษาภาวะตาขี้เกียจ : การรักษาภาวะตาขี้เกียจ โดยการปิดตาข้างที่ดีการผ่าตัดตาเหล่ : พิจารณาการผ่าตัดตามระดับของตาเหล่ ภาวะเส้นประสาทตาเจริญไม่เต็มที่ การมองเห็น และลานสายตาจะไม่เปลี่ยนแปลงเว้นแต่จะมีต้อหิน ร่วมด้วย หลีกเลี่ยงการลดความดันลูกตา อย่างไม่เหมาะสม พยายามปรับปรุงการทำงานของสายตาที่เหลืออยู่ด้วยการแก้ไขค่าสายตาที่เหมาะสมจอประสาทตาลอก ชนิดเซรุ่มที่สัมพันธ์กับคอโลโบมาของจานประสาทตา จอประสาทตาลอก ชนิดมีรอยฉีกขาด ให้พิจารณาการผ่าตัดที่เหมาะสม.
ภาวะเตี้ย : การรักษาด้วยฮอร์โมนการเจริญเติบโต (GH) ผู้ป่วย NS อาจมีภาวะขาด GH 1) อย่างไรก็ตาม ใน NFNS1 การให้ GH มีความเสี่ยงต่อการเพิ่มขนาดของเนื้องอกเส้นประสาท จึงต้องพิจารณาอย่างรอบคอบ 4) .โรคหัวใจ : การขยายบอลลูนหรือการผ่าตัดซ่อมแซมลิ้นหัวใจพัลโมนารีตีบ การติดตามและรักษาด้วยยาสำหรับโรคกล้ามเนื้อหัวใจหนาตัว โรคกล้ามเนื้อหัวใจหนาตัวในเด็กอายุต่ำกว่า 2 ปีมีความเสี่ยงสูงสุดต่อการเสียชีวิตจากโรคหัวใจ5) .ความผิดปกติของเลือด : การเสริมปัจจัยการแข็งตัวของเลือด การประเมินความเสี่ยงต่อการมีเลือดออกก่อนผ่าตัด ในกรณีทารกแรกเกิดของ Tang et al. ภาวะเกล็ดเลือดต่ำที่ดื้อต่อการรักษาเป็นอาการแรก ร่วมกับผนังกั้นหัวใจห้องบนรั่วและน้ำในช่องเยื่อหุ้มปอดชนิด chylothorax 5) .น้ำในช่องเยื่อหุ้มปอดชนิด chylothorax : การระบายน้ำในช่องอก ใน Tian et al. การผ่าตัดด้วยกล้องจุลทรรศน์เพื่อแก้ไขการอุดตันของท่อน้ำเหลืองทรวงอกช่วยให้ภาวะอัลบูมินในเลือดต่ำดีขึ้น 6) .อัณฑะไม่ลงถุง : การผ่าตัดตรึงอัณฑะในเวลาที่เหมาะสม
กลุ่มอาการนูนันไม่มีวิธีการรักษาที่หายขาด การดูแลเป็นแบบประคับประคองตามอาการและติดตามผลระยะยาว
ก่อนผ่าตัด จำเป็นต้องประเมินแนวโน้มการมีเลือดออก (เนื่องจากมักมีภาวะขาดปัจจัยการแข็งตัวของเลือดและความผิดปกติของเกล็ดเลือดร่วมด้วย)
การให้ฮอร์โมนการเจริญเติบโตในผู้ป่วย NFNS1 มีความเสี่ยงต่อการเพิ่มขนาดของเนื้องอกเส้นประสาท จำเป็นต้องพิจารณาอย่างรอบคอบ
Q
การรักษาดวงตาในกลุ่มอาการนูนันทำอย่างไร?
A
การแก้ไขสายตาผิดปกติด้วยแว่นตาหรือคอนแทคเลนส์และการรักษาภาวะตาขี้เกียจ เป็นพื้นฐาน ตาเหล่ พิจารณาผ่าตัดตามระดับความรุนแรง ภาวะเส้นประสาทตา พัฒนาน้อยไม่ลุกลาม หลีกเลี่ยงการลดความดันลูกตา โดยไม่จำเป็น ปรับปรุงการมองเห็น ที่เหลือด้วยการแก้ไขสายตาที่เหมาะสม จอประสาทตาลอก ชนิดเซรุ่มที่เกี่ยวข้องกับคอโลโบมาของจานประสาทตา อาจหายได้เอง ไม่มีการรักษาที่แน่นอน
พยาธิสรีรวิทยาพื้นฐานของกลุ่มอาการนูนันคือความผิดปกติของการควบคุมวิถี RAS-MAPK ทำให้เกิดการส่งสัญญาณอย่างต่อเนื่องและมากเกินไป
PTPN11 (SHP2)
หน้าที่ : ไทโรซีนฟอสฟาเตสชนิดไม่รับสัญญาณ มีกลไกการยับยั้งตัวเองผ่านปฏิสัมพันธ์ภายในโมเลกุลระหว่างโดเมน N-SH2 และโดเมน PTP
ผลของการกลายพันธุ์ NS : กลไกการยับยั้งตัวเองถูกปลดปล่อย ทำให้กิจกรรมฟอสฟาเตสเพิ่มขึ้น (ชนิดเพิ่มหน้าที่) สัญญาณ RAS-MAPK ถูกกระตุ้นมากเกินไป 2)
ความแตกต่างจาก NSML : การกลายพันธุ์ NSML (กลุ่มอาการลีโอพาร์ด) เป็นชนิดลดกิจกรรมของ PTPN11 ซึ่งตรงกันข้ามกับ NS
LZTR1
หน้าที่ : โปรตีนกอลจิในซูเปอร์แฟมิลี BTB-Kelch ส่งเสริมการเกิดโพลิยูบิควิทิเนชันและการย่อยสลายโปรตีเอโซมของ RAS ควบคุมสัญญาณ RAS-MAPK ในทางลบ 3)
การกลายพันธุ์ AD-NS : กระจุกตัวในโดเมน Kelch (พื้นผิวรับรู้ซับสเตรต) เพิ่มสัญญาณ RAS-MAPK (ชนิดเพิ่มหน้าที่) 1) 3)
การกลายพันธุ์ AR-NS : เกิดจากการกลายพันธุ์แบบสูญเสียการทำงาน นอกจากนี้ยังมีหน้าที่จับกับคอมเพล็กซ์ RAF1/SHOC2/PP1CB และส่งเสริมฟอสโฟรีเลชันของ Ser259 ของ RAF1 (ยับยั้งสัญญาณ MAPK)3) .
ส่วนประกอบหลักของเส้นทาง RAS-MAPK มีดังนี้:
ตัวควบคุมเชิงบวก : SHP2 (PTPN11), SOS1, SOS2. การกลายพันธุ์แบบ gain-of-function ในยีนเหล่านี้ทำให้เกิด NS.ปัจจัยส่งเสริมสัญญาณ RAS : MRAS, SHOC2, PPP1CB.MAPK cascade : BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.ปัจจัยควบคุมเชิงลบ : CBL, NF1 , LZTR1, SPRED1, SPRED2. การกลายพันธุ์แบบสูญเสียหน้าที่ทำให้เกิดการเพิ่มสัญญาณ3) .
วิถี RAS-MAPK มีความสำคัญอย่างยิ่งต่อการแบ่งตัวและการเพิ่มจำนวนของเซลล์ และการกลายพันธุ์แบบ gain-of-function ทำให้เกิดการทำงานของเซลล์ที่ผิดปกติในเนื้อเยื่อทั่วร่างกาย ส่งผลให้เกิดฟีโนไทป์ที่ซับซ้อนในหลายอวัยวะ
ในทางจักษุวิทยา ภาวะ hypoplasia ของเส้นประสาทตา เกิดจากการพัฒนาที่ไม่สมบูรณ์ของเซลล์ปมประสาทจอตาและเส้นใยประสาท Coloboma ของจานประสาทตา เกิดจากการปิดรอยแยกของตาไม่สมบูรณ์ โดยมีความชุกประมาณ 3-8 ต่อ 100,000
ในส่วนที่เกี่ยวข้องกับเนื้องอก การกลายพันธุ์ PTPN11 ยังถูกระบุว่าเป็นการกลายพันธุ์แบบ somatic ใน JMML (มะเร็งเม็ดเลือดขาวชนิดไมอีโลโมโนไซติกในเด็ก) MDS และ AML และความเสี่ยงของมะเร็งเม็ดเลือดในวัยเด็กใน NS ประมาณ 3 เท่า 5)
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิกในปัจจุบัน และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
Tian และคณะ (2025) รายงานผู้ป่วย NS ที่มีการกลายพันธุ์ LZTR1 c.850C>T ซึ่งแสดงการเปลี่ยนแปลงเป็นโพรงของหลอดเลือดดำพอร์ทัล, การเจริญไม่สมบูรณ์ของท่อทรวงอก, การขยายของท่อน้ำเหลืองในลำไส้, และภาวะสูญเสียโปรตีนทางลำไส้6) มีการผ่าตัดด้วยกล้องจุลทรรศน์เพื่อแก้ไขการอุดตันของทางออกท่อทรวงอก และระดับอัลบูมินในซีรัมกลับสู่ปกติ การค้นพบนี้ให้ความเข้าใจใหม่เกี่ยวกับพยาธิกำเนิดของความผิดปกติของท่อน้ำเหลือง ที่เกี่ยวข้องกับ NS ที่มีการกลายพันธุ์ LZTR1
Orsolini และคณะ (2024) รายงานชายอายุ 35 ปีที่มีการกลายพันธุ์ LZTR1 c.742G>A โดยไม่มีประวัติอัณฑะไม่ลงถุง แต่มี FSH สูงและภาวะอสุจิน้อย (ความเข้มข้นของอสุจิ 1.5×10⁶/mL)7) ผู้ป่วยรายนี้ชี้ให้เห็นถึงความผิดปกติของต่อมเพศเนื่องจากความเสียหายปฐมภูมิของเซลล์ Sertoli ซึ่งเป็นอิสระจากอัณฑะไม่ลงถุง ซึ่งมีส่วนช่วยในการทำความเข้าใจกลไกภาวะมีบุตรยากในเพศชายใน NS
LZTR1 ยังเป็นยีนที่ก่อให้เกิดโรค schwannomatosis และการวิจัยเกี่ยวกับความทับซ้อนทางคลินิกระหว่าง NS, NF1 และ schwannomatosis กำลังดำเนินไป 3) ความสัมพันธ์ทางชีววิทยาระหว่าง RIT1 และ LZTR1 ก็ได้รับความสนใจเช่นกัน โดยมีการเสนอว่าการกลายพันธุ์ในยีนใดยีนหนึ่งอาจทำให้เกิดการสะสมของ RIT1 และมีส่วนทำให้สัญญาณ MAPK ทำงานมากเกินไป 3) .
สำหรับความสัมพันธ์ระหว่างจีโนไทป์และฟีโนไทป์ การสร้างความสัมพันธ์ดังกล่าวผ่านกลุ่มตัวอย่างขนาดใหญ่ขึ้นเป็นความท้าทายในอนาคต คาดว่าจะมีการระบุปัจจัยทำนายพยากรณ์โรคทางจักษุวิทยาและการพัฒนาเป้าหมายการรักษาที่จำเพาะต่อ NS
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
ถาม AI เกี่ยวกับบทความนี้
คัดลอกข้อความบทความแล้ววางในผู้ช่วย AI ที่คุณต้องการใช้
เปิดผู้ช่วย AI ด้านล่าง แล้ววางข้อความที่คัดลอกลงในช่องแชต