Sindrom Noonan (Noonan syndrome; NS) adalah penyakit genetik (RASopathy) yang disebabkan oleh mutasi gen pada jalur sinyal RAS-MAPK. Pertama kali dilaporkan secara sistematis oleh Jacqueline Noonan pada tahun 1968.
Perkiraan prevalensi adalah 1 dari 1.000 hingga 2.500 kelahiran. Pola pewarisan biasanya autosomal dominan (AD), tetapi sekitar 2/3 kasus disebabkan oleh mutasi baru (de novo). Mutasi LZTR1 yang dapat berupa AD dan AR (autosomal resesif) telah diketahui; pada keluarga Tiongkok yang diteliti oleh Zhao et al., mutasi LZTR1 c.1149+1G>T menunjukkan pewarisan AD 1).
Sindrom Noonan ditandai dengan “ekspresivitas variabel”, di mana fenotipe sangat bervariasi dalam keluarga dengan mutasi yang sama, dan “penetrasi tidak lengkap”, di mana pembawa tidak menunjukkan fenotipe. Han & Park (2024) melaporkan kasus pewarisan paternal dengan mutasi PTPN11 p.Arg498Trp, di mana ayah tidak bergejala atau hanya memiliki disabilitas intelektual ringan, dan 30-40% kerabat yang terkena 2). Mutasi de novo terkait dengan usia ayah lanjut, dan mutasi yang terjadi selama spermatogenesis dianggap memiliki keunggulan selektif.
Secara oftalmologis, sindrom Noonan dapat disertai berbagai kelainan seperti hipoplasia saraf optik, koloboma diskus optikus, kelainan refraksi, dan ptosis.
QSeberapa genetik sindrom Noonan?
A
Biasanya menunjukkan pewarisan autosomal dominan, tetapi sekitar 2/3 kasus disebabkan oleh mutasi de novo (spontan). Secara teoritis, kemungkinan pewarisan dari orang tua yang terkena ke anak adalah 50%, tetapi karena penetrasi tidak lengkap, proporsi gejala yang benar-benar muncul diperkirakan 30-40% 2). Jika ada riwayat keluarga, konseling genetik dianjurkan.
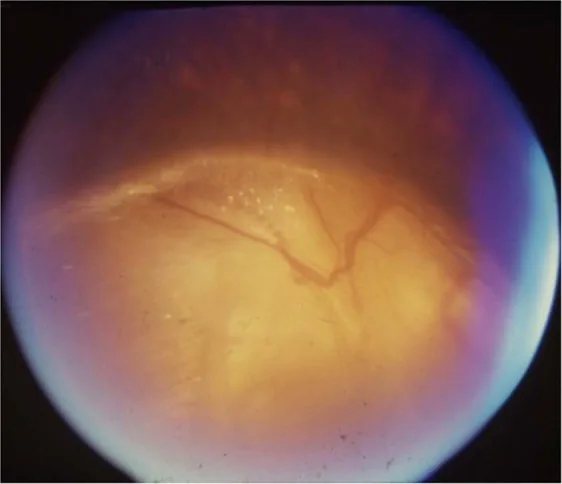
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Gambar yang menunjukkan fitur wajah yang terkait dengan Sindrom Noonan
Wajah khas: Dahi lebar, pangkal hidung cekung, telinga rendah berputar ke belakang dengan heliks tebal. Paling menonjol pada masa bayi dan anak-anak.
Sistem kardiovaskular: Stenosis katup pulmonal (50-60%), kardiomiopati hipertrofik (20%), ASD (6-10%), VSD, limfangiogenesis tidak sempurna.
Kerangka dan Leher: Leher bersayap, dada burung merpati atau dada corong, jarak puting yang lebar, kepala relatif besar.
Hematologis: Perpanjangan PT/PTT, kelainan jumlah dan fungsi trombosit, defisiensi faktor XI (50%). Trombositopenia refrakter pada neonatus dapat menjadi gejala awal 5).
Genitalia: kriptorkismus pada pria (hingga 80%), infertilitas.
Kasus Zhao et al. (anak perempuan 6,6 tahun) menunjukkan strabismus, kelainan refraksi, dan nistagmus, disertai defisiensi hormon pertumbuhan 1). Tian et al. (2025) melaporkan transformasi kavernosa portal, dilatasi limfatik usus, dan enteropati kehilangan protein pada NS dengan mutasi LZTR1, menunjukkan berbagai manifestasi abnormalitas limfatik 6).
Penyebab NS adalah mutasi gain-of-function (atau loss-of-function) pada beberapa gen yang terlibat dalam jalur RAS-MAPK. Frekuensi gen penyebab ditunjukkan di bawah ini.
Gen
Frekuensi
Karakteristik utama
PTPN11
Sekitar 50%
Paling umum. Mutasi gain-of-function dominan
SOS1
Sekitar 10-13%
—
RAF1, RIT1
Masing-masing sekitar 5%
Telah dilaporkan terkait dengan gangguan penglihatan
LZTR1 adalah protein badan Golgi yang termasuk dalam superfamili BTB-Kelch, yang mempromosikan ubiquitinasi dan degradasi RAS untuk mengatur sinyal RAS-MAPK secara negatif 3). Mutasi AD-NS terkonsentrasi pada domain Kelch (permukaan pengenalan substrat) dan meningkatkan sinyal RAS-MAPK 1)3).
Mengenai korelasi antara genotipe dan fenotipe oftalmologis, telah dilaporkan gangguan penglihatan permanen (ketajaman penglihatan terkoreksi terbaik < 0,3) pada pasien dengan mutasi RAF1, KRAS, dan SHOC2, serta ketajaman penglihatan terendah (penglihatan buruk sejak bayi akibat hipoplasia saraf optik) pada pasien dengan mutasi BRAF, namun korelasi pasti belum terbukti dan kohort berukuran kecil (n=25-105).
Pada NFNS (neurofibromatosis-Noonan syndrome) akibat mutasi NF1, sekitar 25% pasien NF1 menunjukkan ciri-ciri mirip Noonan, dan fenotipe kedua penyakit tumpang tindih 4).
QApakah tingkat keparahan gejala mata berbeda tergantung pada jenis gen?
A
Telah dilaporkan gangguan penglihatan permanen pada pasien dengan mutasi RAF1, KRAS, dan SHOC2, serta gangguan penglihatan sejak bayi akibat hipoplasia saraf optik pada pasien dengan mutasi BRAF. Di sisi lain, ada laporan bahwa tidak ditemukan gangguan penglihatan pada pasien dengan mutasi PTPN11. Namun, kohort yang menunjukkan korelasi ini berukuran kecil, dan korelasi genotip-fenotip yang pasti belum ditetapkan saat ini.
Diagnosis NS dilakukan berdasarkan kriteria diagnosis resmi yang didasarkan pada temuan klinis seperti wajah khas, penyakit jantung, dan perawakan pendek, serta dievaluasi oleh spesialis genetika klinis.
Untuk diagnosis pasti, digunakan pemeriksaan genetik molekuler dengan panel gen terkait jalur RAS-MAPK (seperti PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1)2)3). Whole exome sequencing (WES) juga digunakan1)4). Perlu diperhatikan bahwa panel gen NS mungkin tidak menyertakan LZTR13).
NFNS (Neurofibromatosis-Noonan syndrome): Mutasi NF1 menyebabkan tumpang tindih fenotipe NS+NF1. Cari keterlibatan NF1 jika terdapat ≥6 bercak café-au-lait atau ukuran ≥5 mm4). Farncombe dkk. melaporkan neurofibroma pleksiformis pada pasien NS dengan mutasi LZTR1, menunjukkan tumpang tindih klinis dengan NF13).
Operasi strabismus: Operasi dipertimbangkan berdasarkan tingkat strabismus.
Hipoplasia saraf optik: Non-progresif, penglihatan dan lapang pandang tidak berubah kecuali jika disertai glaukoma. Hindari penurunan tekanan intraokular yang sembarangan. Coba perbaiki fungsi visual yang tersisa dengan koreksi refraksi yang tepat.
Ablasio retina serosa terkait koloboma diskus optikus: Tidak ada pengobatan yang pasti, dan beberapa kasus dapat sembuh spontan. Untuk ablasio retina regmatogen, pertimbangkan terapi bedah yang sesuai.
Perawakan pendek: Terapi hormon pertumbuhan (GH). Pasien NS dapat mengalami defisiensi GH 1). Namun, pada NFNS1, pemberian GH berisiko meningkatkan pertumbuhan neurofibroma, sehingga diperlukan pertimbangan yang hati-hati 4).
Penyakit jantung: Dilatasi balon atau perbaikan bedah untuk stenosis katup pulmonal. Observasi dan pengobatan medis untuk kardiomiopati hipertrofik. Kardiomiopati hipertrofik pada anak di bawah 2 tahun merupakan risiko tertinggi kematian jantung5).
Kelainan darah: Penggantian faktor koagulasi. Penilaian risiko perdarahan sebelum operasi. Pada kasus neonatus oleh Tang et al., trombositopenia refrakter merupakan gejala awal, disertai dengan ASD dan kilotoraks 5).
Kilotoraks: Drainase toraks. Pada Tian et al., operasi mikroskopis untuk obstruksi saluran toraks memperbaiki hipoalbuminemia 6).
Kriptorkismus: Orkidopeksi pada waktu yang tepat.
QBagaimana pengobatan mata pada sindrom Noonan?
A
Koreksi refraksi dengan kacamata atau lensa kontak dan terapi ambliopia adalah dasar. Strabismus dipertimbangkan untuk operasi sesuai derajatnya. Hipoplasia saraf optik bersifat non-progresif, hindari penurunan tekanan intraokular yang tidak perlu, dan tingkatkan fungsi visual yang tersisa dengan koreksi refraksi yang tepat. Ablasi retina serosa yang terkait dengan koloboma diskus optikus dapat mengalami resolusi spontan, dan tidak ada pengobatan yang pasti.
Fisiopatologi dasar sindrom Noonan adalah disregulasi jalur RAS-MAPK yang menyebabkan sinyal berkelanjutan dan berlebihan.
PTPN11 (SHP2)
Fungsi: Tirosin fosfatase non-reseptor. Memiliki mekanisme auto-inhibisi melalui interaksi intramolekul antara domain N-SH2 dan domain PTP.
Efek mutasi NS: Mekanisme auto-inhibisi dilepaskan, menyebabkan aktivitas fosfatase meningkat (tipe gain-of-function). Sinyal RAS-MAPK menjadi terlalu aktif 2).
Perbedaan dengan NSML: Mutasi NSML (sindrom leopard) adalah tipe penurunan aktivitas PTPN11, kebalikan dari NS.
LZTR1
Fungsi: Protein Golgi yang termasuk dalam superfamili BTB-Kelch. Mempercepat poliubiquitinasi dan degradasi proteasom RAS, mengatur sinyal RAS-MAPK secara negatif 3).
Mutasi AD-NS: Terkonsentrasi di domain Kelch (permukaan pengenalan substrat). Meningkatkan sinyal RAS-MAPK (tipe gain-of-function) 1)3).
Mutasi AR-NS: Disebabkan oleh mutasi tipe kehilangan fungsi. Juga memiliki fungsi mengikat kompleks RAF1/SHOC2/PP1CB dan mempromosikan fosforilasi Ser259 RAF1 (inaktivasi sinyal MAPK)3).
Komponen utama dari jalur RAS-MAPK adalah sebagai berikut:
Regulator positif: SHP2 (PTPN11), SOS1, SOS2. Mutasi gain-of-function pada gen-gen ini menyebabkan NS.
Faktor promosi sinyal RAS: MRAS, SHOC2, PPP1CB.
Kaskade MAPK: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Faktor regulasi negatif: CBL, NF1, LZTR1, SPRED1, SPRED2. Mutasi kehilangan fungsi menyebabkan peningkatan sinyal3).
Jalur RAS-MAPK sangat penting untuk diferensiasi dan proliferasi sel, dan mutasi gain-of-function menyebabkan fungsi sel abnormal di jaringan seluruh tubuh, menghasilkan fenotip kompleks yang melibatkan banyak organ.
Secara oftalmologis, hipoplasia saraf optik disebabkan oleh perkembangan sel ganglion retina dan serabut saraf yang tidak sempurna. Koloboma diskus optikus disebabkan oleh penutupan fisura optik yang tidak sempurna, dengan prevalensi diperkirakan 3-8 per 100.000.
Terkait tumor, mutasi PTPN11 juga diidentifikasi sebagai mutasi somatik pada JMML (leukemia mielomonositik juvenil), MDS, dan AML, dan risiko keganasan hematologi pada masa kanak-kanak pada NS diperkirakan 3 kali lipat 5).
7. Penelitian Terbaru dan Prospek Masa Depan (Laporan Tahap Penelitian)
Tian et al. (2025) melaporkan kasus NS dengan mutasi LZTR1 c.850C>T yang menunjukkan transformasi kavernosa vena porta, hipoplasia duktus toraks, limfangiektasia usus, dan enteropati kehilangan protein6). Operasi mikroskopis dilakukan untuk obstruksi outlet duktus toraks, dan kadar albumin serum menjadi normal. Temuan ini memberikan wawasan baru tentang patogenesis limfangiogenesis abnormal yang terkait dengan NS mutasi LZTR1.
Orsolini et al. (2024) melaporkan seorang pria berusia 35 tahun dengan mutasi LZTR1 c.742G>A, tanpa riwayat kriptorkismus, namun menunjukkan FSH tinggi dan oligozoospermia (konsentrasi sperma 1,5×10⁶/mL)7). Kasus ini menunjukkan adanya disfungsi gonad akibat kerusakan sel Sertoli primer yang independen dari kriptorkismus, berkontribusi pada pemahaman mekanisme infertilitas pria pada NS.
LZTR1 juga merupakan gen penyebab schwannomatosis, dan penelitian tentang tumpang tindih klinis antara NS, NF1, dan schwannomatosis sedang berkembang 3). Hubungan biologis antara RIT1 dan LZTR1 juga menjadi perhatian, di mana mutasi pada salah satu dari keduanya diduga menyebabkan akumulasi RIT1 dan berkontribusi pada hiperaktivasi sinyal MAPK 3).
Mengenai korelasi genotipe-fenotipe, penetapannya melalui kohort yang lebih besar merupakan tantangan di masa depan. Identifikasi faktor prediktif prognosis oftalmologis dan pengembangan target terapi spesifik NS diharapkan dapat tercapai.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.