A síndrome de Noonan (Noonan syndrome; NS) é uma doença genética (RASopatia) causada por mutações em genes da via de sinalização RAS-MAPK. Foi descrita sistematicamente pela primeira vez por Jacqueline Noonan em 1968.
A prevalência estimada é de 1 em cada 1.000 a 2.500 nascimentos. O padrão de herança é geralmente autossômico dominante (AD), mas cerca de 2/3 dos casos são devidos a mutações de novo. Mutações no LZTR1 que podem ser tanto AD quanto AR (autossômico recessivo) são conhecidas; na família chinesa estudada por Zhao et al., a mutação LZTR1 c.1149+1G>T mostrou herança AD 1).
A síndrome de Noonan é caracterizada por “expressividade variável”, onde os fenótipos variam amplamente dentro de famílias com a mesma mutação, e “penetrância incompleta”, onde portadores não apresentam fenótipo. Han & Park (2024) relataram um caso de herança paterna com mutação PTPN11 p.Arg498Trp, onde o pai era assintomático ou apresentava apenas deficiência intelectual leve, e 30-40% dos parentes afetados 2). Mutações de novo estão associadas à idade paterna avançada, e acredita-se que mutações ocorridas durante a espermatogênese tenham vantagem seletiva.
Geralmente apresenta herança autossômica dominante, mas cerca de 2/3 dos casos são devidos a mutações de novo (espontâneas). Teoricamente, a probabilidade de transmissão de um pai afetado para o filho é de 50%, mas devido à penetrância incompleta, a proporção real de manifestação dos sintomas é de 30 a 40% 2). Se houver histórico familiar, o aconselhamento genético é recomendado.
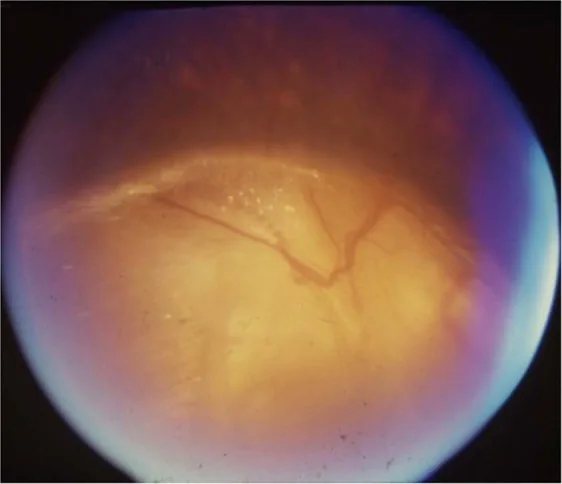
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Imagem mostrando características faciais associadas à Síndrome de Noonan
Aparência facial característica: testa larga, ponte nasal deprimida, orelhas baixas e rodadas posteriormente com hélice espessa. Mais proeminente na infância e na primeira infância.
Sistema cardiovascular: Estenose da válvula pulmonar (50-60%), cardiomiopatia hipertrófica (20%), CIA (6-10%), CIV, linfangiogênese incompleta.
Esqueleto e Pescoço: Pescoço alado, tórax em quilha ou tórax em funil, hipertelorismo mamário, macrocefalia relativa.
Hematológicos: prolongamento do TP/TTPA, alterações na contagem e função plaquetárias, deficiência do fator XI (50%). A trombocitopenia refratária no período neonatal pode ser o primeiro sintoma 5).
Genitália: criptorquidia em homens (até 80%), infertilidade.
O caso de Zhao et al. (menina de 6,6 anos) apresentou estrabismo, erro refrativo e nistagmo, com deficiência de hormônio do crescimento 1). Tian et al. (2025) relataram transformação cavernosa portal, dilatação linfática intestinal e enteropatia perdedora de proteínas na SN com mutação LZTR1, demonstrando diversas manifestações de anormalidades linfáticas 6).
QQuais são os sintomas oculares da síndrome de Noonan?
A causa da SN são mutações de ganho (ou perda) de função em vários genes envolvidos na via RAS-MAPK. A frequência dos genes causadores é mostrada abaixo.
Gene
Frequência
Principais características
PTPN11
Cerca de 50%
Mais comum. Mutações de ganho de função são predominantes
SOS1
Cerca de 10-13%
—
RAF1, RIT1
Cerca de 5% cada
Relato de associação com deficiência visual
LZTR1
cerca de 8%
Padrões de herança AD e AR
KRAS, BRAF
cada um <5%
Mutações BRAF relatadas com pior acuidade visual
LZTR1 é uma proteína do aparelho de Golgi pertencente à superfamília BTB-Kelch, que promove a ubiquitinação e degradação de RAS para regular negativamente a sinalização RAS-MAPK 3). Mutações AD-NS concentram-se no domínio Kelch (superfície de reconhecimento do substrato) e aumentam a sinalização RAS-MAPK 1)3).
Quanto à correlação entre genótipo e fenótipo oftalmológico, foram relatados comprometimento visual permanente (melhor acuidade visual corrigida < 0,3) em pacientes com mutações RAF1, KRAS e SHOC2, e pior acuidade visual (má visão desde a infância devido a hipoplasia do nervo óptico) em pacientes com mutações BRAF, mas a correlação definitiva não foi estabelecida e as coortes são pequenas (n=25-105).
Na NFNS (neurofibromatose-síndrome de Noonan) devido à mutação NF1, cerca de 25% dos pacientes com NF1 apresentam características semelhantes a Noonan, e os fenótipos das duas doenças se sobrepõem 4).
QA gravidade dos sintomas oculares varia de acordo com o tipo de gene?
A
Foram relatadas deficiências visuais permanentes em pacientes com mutações RAF1, KRAS e SHOC2, e deficiência visual desde a infância devido à hipoplasia do nervo óptico em pacientes com mutação BRAF. Por outro lado, há relatos de que nenhuma deficiência visual foi observada em pacientes com mutação PTPN11. No entanto, as coortes que mostram essas correlações são de pequena escala e, atualmente, não foi estabelecida uma correlação genótipo-fenótipo definitiva.
O diagnóstico de SN é realizado de acordo com critérios diagnósticos formais baseados em achados clínicos como fácies característica, cardiopatia e baixa estatura, sendo avaliado por um especialista em genética clínica.
Para o diagnóstico definitivo, utiliza-se o exame genético molecular com um painel de genes relacionados à via RAS-MAPK (como PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1)2)3). O sequenciamento do exoma completo (WES) também é utilizado1)4). Deve-se observar que o painel genético da SN pode não incluir LZTR13).
NFNS (Neurofibromatose-Síndrome de Noonan): Mutação NF1 causa sobreposição de fenótipos NS+NF1. Pesquise associação com NF1 se ≥6 manchas café-com-leite ou tamanho ≥5 mm4). Farncombe et al. relataram neurofibroma plexiforme em pacientes NS com mutação LZTR1, indicando sobreposição clínica com NF13).
Diagnóstico diferencial de coloboma de papila óptica: Estafiloma peripapilar, síndrome da glória matinal, PFV/PHPV papilar, macropapila.
Outras síndromes: Síndrome CHARGE, síndrome de Turner, síndrome cardiofaciocutânea (CFC), síndrome de Costello, síndrome de Legius.
Hipoplasia do nervo óptico: Não progressiva, a visão e o campo visual não mudam a menos que haja glaucoma associado. Evite reduzir a pressão intraocular indiscriminadamente. Tente melhorar a função visual residual com correção refrativa adequada.
Descolamento seroso da retina associado a coloboma do disco óptico: Não há tratamento definido, e alguns casos podem regredir espontaneamente. Para descolamento regmatogênico, considere cirurgia apropriada.
Baixa estatura: Terapia com hormônio do crescimento (GH). Pacientes com NS podem apresentar deficiência de GH 1). No entanto, na NFNS1, a administração de GH apresenta risco de aumento de neurofibromas, exigindo avaliação cuidadosa 4).
Doenças cardíacas: Dilatação por balão ou reparo cirúrgico para estenose da válvula pulmonar. Acompanhamento e tratamento medicamentoso para cardiomiopatia hipertrófica. A cardiomiopatia hipertrófica em crianças com menos de 2 anos é o maior risco de morte cardíaca5).
Anormalidades sanguíneas: Reposição de fatores de coagulação. Avaliação do risco de sangramento pré-operatório. No caso neonatal de Tang et al., a trombocitopenia refratária foi o sintoma inicial, associada a CIA e quilotórax 5).
Quilotórax: Drenagem torácica. Em Tian et al., a cirurgia microscópica para obstrução da saída do ducto torácico melhorou a hipoalbuminemia 6).
Criptorquidia: Orquidopexia no momento adequado.
QComo é feito o tratamento ocular da síndrome de Noonan?
A
A correção refrativa com óculos ou lentes de contato e o tratamento da ambliopia são a base. O estrabismo é considerado para cirurgia conforme o grau. A hipoplasia do nervo óptico é não progressiva; evite redução desnecessária da pressão intraocular e melhore a função visual residual com correção refrativa adequada. O descolamento seroso da retina associado ao coloboma do disco óptico pode regredir espontaneamente, e não há tratamento definido.
6. Fisiopatologia e Mecanismo Detalhado de Patogênese
A fisiopatologia fundamental da síndrome de Noonan é a desregulação da via RAS-MAPK, resultando em sinalização contínua e excessiva.
PTPN11 (SHP2)
Função: Tirosina fosfatase não receptora. Possui mecanismo de autoinibição por interação intramolecular entre o domínio N-SH2 e o domínio PTP.
Efeito das mutações NS: O mecanismo de autoinibição é liberado, aumentando a atividade da fosfatase (tipo ganho de função). A sinalização RAS-MAPK fica hiperativada 2).
Diferença da NSML: Mutações NSML (síndrome de leopard) são do tipo de perda de atividade da PTPN11, opostas à NS.
LZTR1
Função: Proteína do Golgi pertencente à superfamília BTB-Kelch. Promove a poliubiquitinação e degradação proteassômica de RAS, regulando negativamente a sinalização RAS-MAPK 3).
Mutações AD-NS: Concentram-se no domínio Kelch (superfície de reconhecimento do substrato). Aumentam a sinalização RAS-MAPK (tipo ganho de função) 1)3).
Mutação AR-NS: Causada por mutações do tipo perda de função. Também tem a função de se ligar ao complexo RAF1/SHOC2/PP1CB e promover a fosforilação da Ser259 do RAF1 (inativação da sinalização MAPK)3).
Os principais componentes da via RAS-MAPK são os seguintes:
Reguladores positivos: SHP2 (PTPN11), SOS1, SOS2. Mutações de ganho de função nesses genes causam a SN.
Fatores promotores da sinalização RAS: MRAS, SHOC2, PPP1CB.
Cascata MAPK: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Reguladores negativos: CBL, NF1, LZTR1, SPRED1, SPRED2. Mutações de perda de função causam aumento da sinalização3).
A via RAS-MAPK é crucial para a diferenciação e proliferação celular, e mutações de ganho de função causam função celular anormal em tecidos de todo o corpo, resultando em um fenótipo complexo multiorgânico.
Oftalmologicamente, a hipoplasia do nervo óptico é devida ao desenvolvimento incompleto das células ganglionares da retina e fibras nervosas. O coloboma do disco óptico resulta do fechamento incompleto da fissura óptica, com prevalência estimada de 3-8 por 100.000.
Em relação a tumores, mutações PTPN11 também são identificadas como mutações somáticas em JMML (leucemia mielomonocítica juvenil), MDS e LMA, e o risco de neoplasias hematológicas na infância na SN é estimado em 3 vezes 5).
7. Pesquisas Recentes e Perspectivas Futuras (Relatos em Fase de Pesquisa)
Tian et al. (2025) relataram um caso de SN com mutação LZTR1 c.850C>T apresentando transformação cavernosa da veia porta, hipoplasia do ducto torácico, linfangiectasia intestinal e enteropatia perdedora de proteínas6). Foi realizada cirurgia microscópica para obstrução da saída do ducto torácico, com normalização da albumina sérica. Esses achados fornecem novos insights sobre a patogênese das anomalias linfáticas associadas à SN com mutação LZTR1.
Orsolini et al. (2024) relataram um homem de 35 anos com mutação LZTR1 c.742G>A, sem história de criptorquidia, mas apresentando FSH elevado e oligozoospermia (concentração de esperma 1,5×10⁶/mL)7). Este caso sugere disfunção gonadal devido a dano primário das células de Sertoli, independente de criptorquidia, contribuindo para a compreensão do mecanismo de infertilidade masculina na SN.
LZTR1 também é o gene causador da schwannomatose, e pesquisas sobre a sobreposição clínica entre SN, NF1 e schwannomatose estão em andamento 3). A relação biológica entre RIT1 e LZTR1 também tem recebido atenção, sugerindo que mutações em qualquer um deles podem levar ao acúmulo de RIT1 e contribuir para a hiperativação da via MAPK 3).
Quanto à correlação genótipo-fenótipo, seu estabelecimento por meio de coortes maiores é um desafio futuro. Espera-se a identificação de fatores preditivos de prognóstico oftalmológico e o desenvolvimento de alvos terapêuticos específicos para SN.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.