Hội chứng Noonan (Noonan syndrome; NS) là bệnh di truyền (RASopathy) do đột biến gen trong con đường tín hiệu RAS-MAPK. Lần đầu tiên được Jacqueline Noonan báo cáo một cách có hệ thống vào năm 1968.
Tỷ lệ mắc ước tính khoảng 1 trên 1.000 đến 2.500 ca sinh. Kiểu di truyền thường là trội trên nhiễm sắc thể thường (AD), nhưng khoảng 2/3 trường hợp là do đột biến mới (de novo). Các đột biến LZTR1 có thể theo cả hai dạng AD và AR (lặn trên nhiễm sắc thể thường) đã được biết đến; trong gia đình người Trung Quốc được nghiên cứu bởi Zhao và cộng sự, đột biến LZTR1 c.1149+1G>T cho thấy di truyền AD 1).
Hội chứng Noonan được đặc trưng bởi “tính biểu hiện thay đổi”, trong đó kiểu hình khác nhau đáng kể trong cùng một gia đình có cùng đột biến, và “tính thâm nhập không hoàn toàn”, trong đó người mang gen không biểu hiện kiểu hình. Han & Park (2024) báo cáo một trường hợp di truyền từ cha với đột biến PTPN11 p.Arg498Trp, trong đó người cha không có triệu chứng hoặc chỉ bị khuyết tật trí tuệ nhẹ, và 30-40% người thân bị ảnh hưởng 2). Đột biến de novo liên quan đến tuổi cha cao, và các đột biến xảy ra trong quá trình sinh tinh được cho là có lợi thế chọn lọc.
Về mặt nhãn khoa, hội chứng Noonan có thể kèm theo nhiều bất thường như thiểu sản thần kinh thị giác, u nhú thị giác dạng coloboma, tật khúc xạ và sụp mi.
QHội chứng Noonan di truyền như thế nào?
A
Thông thường di truyền trội trên nhiễm sắc thể thường, nhưng khoảng 2/3 trường hợp là do đột biến mới (tự phát). Về mặt lý thuyết, xác suất di truyền từ cha mẹ bị bệnh sang con là 50%, nhưng do thâm nhập không hoàn toàn, tỷ lệ thực tế xuất hiện triệu chứng được ước tính là 30-40% 2). Nếu có tiền sử gia đình, nên tư vấn di truyền.
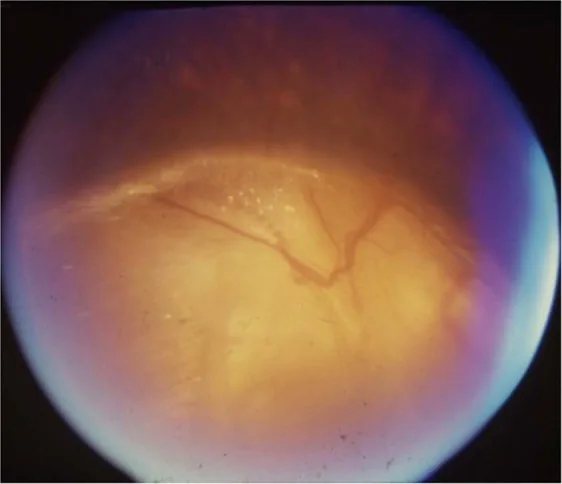
Gopal Lingam, Alok C Sen, Vijaya Lingam et al. Ocular coloboma—a comprehensive review for the clinician. Eye. 2021 Mar 21; 35(8):2086. Figure 3. PMCID: PMC8302742. License: CC BY.
Hình ảnh cho thấy các đặc điểm khuôn mặt liên quan đến Hội chứng Noonan
Tật khúc xạ: Cận thị, viễn thị (bao gồm nặng) và loạn thị. Gặp ở hầu hết bệnh nhân.
Dấu hiệu mắt ngoài: Sụp mi, khoảng cách hai mắt xa (hypertelorism), khe mi chếch xuống (down-slanting palpebral fissures), lông mày cong cao, và nếp gấp góc trong (epicanthus).
Dấu hiệu đoạn trước: Giác mạc hình nón, dây thần kinh giác mạc nổi rõ, mống mắt xanh/xanh lục, vòng phôi sau (posterior embryotoxon), và đục thủy tinh thể.
Đặc điểm khuôn mặt: Trán rộng, sống mũi lõm, tai thấp và xoay ra sau với vành tai dày. Nổi bật nhất ở giai đoạn sơ sinh và trẻ nhỏ.
Hệ tim mạch: Hẹp van động mạch phổi (50-60%), bệnh cơ tim phì đại (20%), thông liên nhĩ (6-10%), thông liên thất, bất thường phát triển bạch mạch.
Xương và cổ: Cổ có màng, ngực ức gà hoặc ngực phễu, khoảng cách núm vú rộng, đầu to tương đối.
Huyết học: Kéo dài PT/PTT, bất thường số lượng và chức năng tiểu cầu, thiếu hụt yếu tố XI (50%). Giảm tiểu cầu kháng trị ở trẻ sơ sinh có thể là triệu chứng khởi phát 5).
Cơ quan sinh dục: tinh hoàn ẩn ở nam (lên đến 80%), vô sinh.
Trường hợp của Zhao et al. (bé gái 6,6 tuổi) có lác, tật khúc xạ và rung giật nhãn cầu, kèm thiếu hụt hormone tăng trưởng 1). Tian et al. (2025) báo cáo biến đổi hang vị cửa, giãn bạch mạch ruột và bệnh ruột mất protein trong NS có đột biến LZTR1, cho thấy nhiều biểu hiện bất thường bạch mạch 6).
QCác triệu chứng về mắt của hội chứng Noonan là gì?
A
Biểu hiện đa dạng bao gồm tật khúc xạ (cận thị, viễn thị, loạn thị), sụp mi, lệch mắt (lác), rung giật nhãn cầu và nhược thị. Các biểu hiện nặng hơn có thể gồm giảm sản thần kinh thị, u nhú thị giác và giác mạc hình chóp. Khuyến cáo đánh giá mắt toàn diện khi chẩn đoán.
Nguyên nhân của NS là đột biến tăng chức năng (hoặc giảm chức năng) ở nhiều gen tham gia con đường RAS-MAPK. Tần số các gen gây bệnh được trình bày dưới đây.
Gen
Tần suất
Đặc điểm chính
PTPN11
Khoảng 50%
Phổ biến nhất. Đột biến tăng chức năng chiếm ưu thế
SOS1
Khoảng 10-13%
—
RAF1, RIT1
Mỗi loại khoảng 5%
Đã có báo cáo về mối liên quan với suy giảm thị lực
LZTR1
khoảng 8%
Kiểu di truyền AD và AR
KRAS, BRAF
mỗi loại <5%
Đột biến BRAF được báo cáo liên quan thị lực thấp nhất
LZTR1 là protein bộ Golgi thuộc siêu họ BTB-Kelch, thúc đẩy quá trình ubiquitin hóa và phân hủy RAS để điều hòa tiêu cực tín hiệu RAS-MAPK 3). Các đột biến AD-NS tập trung ở vùng Kelch (bề mặt nhận diện cơ chất) và làm tăng tín hiệu RAS-MAPK 1)3).
Về mối tương quan giữa kiểu gen và kiểu hình nhãn khoa, đã có báo cáo về suy giảm thị lực vĩnh viễn (thị lực tốt nhất sau điều chỉnh < 0,3) ở bệnh nhân có đột biến RAF1, KRAS và SHOC2, và thị lực thấp nhất (thị lực kém từ sơ sinh do thiểu sản thần kinh thị giác) ở bệnh nhân có đột biến BRAF, nhưng mối tương quan chắc chắn chưa được thiết lập và cỡ mẫu nhỏ (n=25-105).
Trong NFNS (u xơ thần kinh-hội chứng Noonan) do đột biến NF1, khoảng 25% bệnh nhân NF1 có biểu hiện đặc điểm giống Noonan, và kiểu hình của hai bệnh chồng lấn nhau 4).
QMức độ nghiêm trọng của triệu chứng mắt có thay đổi tùy theo loại gen không?
A
Đã có báo cáo về suy giảm thị lực vĩnh viễn ở bệnh nhân có đột biến RAF1, KRAS và SHOC2, và suy giảm thị lực từ giai đoạn sơ sinh do giảm sản thần kinh thị giác ở bệnh nhân có đột biến BRAF. Mặt khác, có báo cáo cho rằng không phát hiện suy giảm thị lực ở bệnh nhân có đột biến PTPN11. Tuy nhiên, các đoàn hệ cho thấy mối tương quan này có quy mô nhỏ và hiện tại chưa thiết lập được mối tương quan kiểu gen-kiểu hình xác định.
Chẩn đoán NS được thực hiện theo các tiêu chuẩn chẩn đoán chính thức dựa trên các phát hiện lâm sàng như khuôn mặt đặc trưng, bệnh tim và chiều cao thấp, và được đánh giá bởi bác sĩ chuyên khoa di truyền lâm sàng.
Để chẩn đoán xác định, sử dụng xét nghiệm di truyền phân tử với bảng gen liên quan đến con đường RAS-MAPK (như PTPN11, SOS1, RAF1, RIT1, KRAS, LZTR1)2)3). Giải trình tự toàn bộ exome (WES) cũng được sử dụng1)4). Cần lưu ý rằng bảng gen NS có thể không bao gồm LZTR13).
NFNS (Hội chứng u xơ thần kinh-Noonan): Đột biến NF1 gây chồng lấn kiểu hình NS+NF1. Tìm kiếm kết hợp NF1 nếu có ≥6 đốm café-au-lait hoặc kích thước ≥5 mm4). Farncombe và cộng sự báo cáo u xơ thần kinh đám rối ở bệnh nhân NS có đột biến LZTR1, cho thấy chồng lấn lâm sàng với NF13).
Chẩn đoán phân biệt u nguyên bào thần kinh thị giác: U nhú quanh gai thị, hội chứng morning glory, PFV/PHPV ở gai thị, gai thị to.
Các hội chứng khác: Hội chứng CHARGE, hội chứng Turner, hội chứng tim-mặt-da (CFC), hội chứng Costello, hội chứng Legius.
Chỉnh khúc xạ: Chỉnh tật khúc xạ bằng kính mắt hoặc kính áp tròng.
Điều trị nhược thị: Điều trị nhược thị bằng cách che mắt lành.
Phẫu thuật lác: Phẫu thuật được xem xét tùy theo mức độ lác.
Thiểu sản thần kinh thị giác: Không tiến triển, thị lực và thị trường không thay đổi trừ khi kèm glôcôm. Tránh hạ nhãn áp tùy tiện. Cố gắng cải thiện chức năng thị giác còn lại bằng chỉnh khúc xạ phù hợp.
Bong võng mạc thanh dịch liên quan đến u nhú thị giác: Không có phương pháp điều trị xác định, một số trường hợp có thể tự thoái lui. Đối với bong võng mạc do rách, cân nhắc phẫu thuật thích hợp.
Chiều cao thấp: Liệu pháp hormone tăng trưởng (GH). Bệnh nhân NS có thể bị thiếu hụt GH 1). Tuy nhiên, ở NFNS1, dùng GH có nguy cơ làm tăng kích thước u xơ thần kinh, cần cân nhắc cẩn thận 4).
Bệnh tim: Nong bóng hoặc phẫu thuật sửa chữa hẹp van động mạch phổi. Theo dõi và điều trị bằng thuốc cho bệnh cơ tim phì đại. Bệnh cơ tim phì đại ở trẻ dưới 2 tuổi là nguy cơ tử vong do tim cao nhất5).
Bất thường máu: Bổ sung yếu tố đông máu. Đánh giá nguy cơ chảy máu trước phẫu thuật. Trong trường hợp sơ sinh của Tang et al., giảm tiểu cầu kháng trị là triệu chứng đầu tiên, kèm theo thông liên nhĩ và tràn dịch dưỡng chấp 5).
Tràn dịch dưỡng chấp: Dẫn lưu khoang ngực. Trong nghiên cứu của Tian et al., phẫu thuật vi thể để giải quyết tắc nghẽn ống ngực đã cải thiện tình trạng giảm albumin máu 6).
Tinh hoàn ẩn: Phẫu thuật hạ tinh hoàn đúng thời điểm.
QĐiều trị mắt cho hội chứng Noonan được thực hiện như thế nào?
A
Chỉnh khúc xạ bằng kính mắt hoặc kính áp tròng và điều trị nhược thị là cơ bản. Lác được xem xét phẫu thuật tùy theo mức độ. Thiểu sản thần kinh thị giác không tiến triển, tránh hạ nhãn áp không cần thiết, cải thiện chức năng thị giác còn lại bằng chỉnh khúc xạ phù hợp. Bong võng mạc thanh dịch kèm theo u nhú thị giác dạng coloboma có thể tự thoái lui, không có phương pháp điều trị xác định.
Sinh lý bệnh cơ bản của hội chứng Noonan là rối loạn điều hòa con đường RAS-MAPK dẫn đến tín hiệu liên tục và quá mức.
PTPN11 (SHP2)
Chức năng: Tyrosine phosphatase không thụ thể. Có cơ chế tự ức chế thông qua tương tác nội phân tử giữa miền N-SH2 và miền PTP.
Ảnh hưởng của đột biến NS: Cơ chế tự ức chế bị giải phóng, làm tăng hoạt tính phosphatase (dạng tăng chức năng). Tín hiệu RAS-MAPK bị hoạt hóa quá mức 2).
Khác biệt với NSML: Đột biến NSML (hội chứng leopard) là dạng giảm hoạt tính của PTPN11, trái ngược với NS.
LZTR1
Chức năng: Protein Golgi thuộc siêu họ BTB-Kelch. Thúc đẩy quá trình polyubiquitin hóa và phân giải proteasome của RAS, điều hòa tiêu cực tín hiệu RAS-MAPK 3).
Đột biến AD-NS: Tập trung ở miền Kelch (bề mặt nhận diện cơ chất). Làm tăng tín hiệu RAS-MAPK (dạng tăng chức năng) 1)3).
Đột biến AR-NS: Do đột biến mất chức năng. Cũng có chức năng liên kết với phức hợp RAF1/SHOC2/PP1CB và thúc đẩy quá trình phosphoryl hóa Ser259 của RAF1 (bất hoạt tín hiệu MAPK)3).
Các thành phần chính của con đường RAS-MAPK như sau:
Các yếu tố điều hòa dương: SHP2 (PTPN11), SOS1, SOS2. Các đột biến tăng chức năng ở các gen này gây ra NS.
Các yếu tố thúc đẩy tín hiệu RAS: MRAS, SHOC2, PPP1CB.
Thác MAPK: BRAF, RAF1, MAP2K1, MAP2K2, MAPK1.
Các yếu tố điều hòa âm tính: CBL, NF1, LZTR1, SPRED1, SPRED2. Các đột biến mất chức năng gây tăng tín hiệu3).
Con đường RAS-MAPK rất quan trọng cho sự biệt hóa và tăng sinh tế bào, và các đột biến tăng chức năng gây ra chức năng tế bào bất thường trong các mô toàn thân, dẫn đến kiểu hình phức tạp liên quan nhiều cơ quan.
Về mặt nhãn khoa, thiểu sản thần kinh thị giác là do sự phát triển không hoàn chỉnh của các tế bào hạch võng mạc và sợi thần kinh. U nguyên bào thần kinh thị giác (coloboma đĩa thị) là do đóng không hoàn toàn khe thị, với tỷ lệ mắc ước tính 3-8 trên 100.000.
Liên quan đến khối u, đột biến PTPN11 cũng được xác định là đột biến soma trong JMML (bệnh bạch cầu tủy bào đơn nhân thiếu niên), MDS và AML, và nguy cơ ung thư huyết học thời thơ ấu trong NS được ước tính gấp 3 lần 5).
7. Nghiên cứu mới nhất và triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
Tian và cộng sự (2025) báo cáo một trường hợp NS có đột biến LZTR1 c.850C>T biểu hiện biến dạng hang tĩnh mạch cửa, giảm sản ống ngực, giãn bạch huyết ruột và bệnh lý ruột mất protein6). Phẫu thuật vi phẫu được thực hiện để giải quyết tắc nghẽn đầu ra ống ngực, và albumin huyết thanh trở về bình thường. Những phát hiện này cung cấp hiểu biết mới về cơ chế bệnh sinh của bất thường bạch huyết liên quan đến NS đột biến LZTR1.
Orsolini và cộng sự (2024) báo cáo một nam giới 35 tuổi có đột biến LZTR1 c.742G>A, không có tiền sử tinh hoàn ẩn, nhưng có FSH cao và thiểu tinh (nồng độ tinh trùng 1,5×10⁶/mL)7). Trường hợp này gợi ý rối loạn chức năng sinh dục do tổn thương tế bào Sertoli nguyên phát, độc lập với tinh hoàn ẩn, góp phần làm sáng tỏ cơ chế vô sinh nam trong NS.
Mối tương quan kiểu gen-kiểu hình và thách thức trong tương lai
LZTR1 cũng là gen gây bệnh u bao sợi thần kinh (schwannomatosis), và nghiên cứu về sự chồng chéo lâm sàng giữa NS, NF1 và schwannomatosis đang tiến triển 3). Mối quan hệ sinh học giữa RIT1 và LZTR1 cũng được chú ý, với giả thuyết rằng đột biến ở một trong hai gen có thể gây tích tụ RIT1 và góp phần hoạt hóa quá mức tín hiệu MAPK 3).
Về mối tương quan kiểu gen - kiểu hình, việc thiết lập nó thông qua các đoàn hệ lớn hơn là thách thức trong tương lai. Việc xác định các yếu tố dự báo tiên lượng nhãn khoa và phát triển các mục tiêu điều trị đặc hiệu cho NS được kỳ vọng.
Zhao X, Li Z, Wang L, Lan Z, Lin F, Zhang W, et al. A Chinese family with Noonan syndrome caused by a heterozygous variant in LZTR1: a case report and literature review. BMC endocrine disorders. 2021;21(1):2. doi:10.1186/s12902-020-00666-6. PMID:33407364; PMCID:PMC7788825.
Han JY, Park J. Paternally Inherited Noonan Syndrome Caused by a PTPN11 Variant May Exhibit Mild Symptoms: A Case Report and Literature Review. Genes. 2024;15(4). doi:10.3390/genes15040445. PMID:38674380; PMCID:PMC11050143.
Farncombe KM, Thain E, Barnett-Tapia C, Sadeghian H, Kim RH. LZTR1 molecular genetic overlap with clinical implications for Noonan syndrome and schwannomatosis. BMC medical genomics. 2022;15(1):160. doi:10.1186/s12920-022-01304-x. PMID:35840934; PMCID:PMC9288044.
Dalili S, Hoseini Nouri SA, Bayat R, Koohmanaee S, Tabrizi M, Zarkesh M, Tarang A, Mahdieh N. Neurofibromatosis-Noonan syndrome and growth deficiency in an Iranian girl due to a pathogenic variant in NF1 gene. Hum Genomics. 2023;17(1):12. doi:10.1186/s40246-023-00460-0.
Tang X, Chen Z, Shen X, Xie T, Wang X, Liu T, et al. Refractory thrombocytopenia could be a rare initial presentation of Noonan syndrome in newborn infants: a case report and literature review. BMC pediatrics. 2022;22(1):142. doi:10.1186/s12887-021-02909-4. PMID:35300644; PMCID:PMC8928670.
Tian QJ, Zhang LJ, Zhang Q, Liu FC, Xie M, Cai JZ, et al. Protein-losing enteropathy and multiple vasculature dysplasia in LZTR1-related Noonan syndrome: A case report and review of literature. World journal of gastroenterology. 2025;31(17):105347. doi:10.3748/wjg.v31.i17.105347. PMID:40521264; PMCID:PMC12159983.
Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M, Canale D. Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front Endocrinol. 2024;15:1354699. doi:10.3389/fendo.2024.1354699.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.