El síndrome de Weill-Marchesani (WMS) es un trastorno hereditario del tejido conectivo. También se denomina síndrome de esferofaquia-braquimorfia o distrofia mesodérmica. Otros nombres incluyen síndrome de Marchesani y síndrome de Marfan invertido. Fue descrito por primera vez por Weill y Marchesani hace más de 80 años. 2)
La prevalencia se estima en 1 caso por cada 100,000 personas. 1) La herencia puede ser autosómica recesiva (AR) o autosómica dominante (AD), con un 45% autosómica recesiva, 39% autosómica dominante y 16% esporádica. 1)
Se clasifica en cuatro subtipos según el gen causante.
WMS1: ADAMTS10 (autosómica recesiva) 2)
WMS2: FBN1 (autosómica dominante)
WMS3: LTBP2 (autosómica recesiva) 1)
WMS4: ADAMTS17 (autosómica recesiva)
Se caracteriza por presentar un tipo de cuerpo opuesto al síndrome de Marfan, y también se denomina “síndrome de Marfan invertido”. Mientras que el síndrome de Marfan muestra estatura alta, aracnodactilia y luxación del cristalino hacia arriba, el síndrome de Weill-Marchesani muestra estatura baja, braquidactilia y luxación del cristalino hacia abajo.
Q¿Qué tan raro es el síndrome de Weill-Marchesani?
A
Es una enfermedad rara con una prevalencia estimada de 1 caso por cada 100,000 personas. 1) El patrón de herencia es autosómico recesivo en el 45%, autosómico dominante en el 39% y esporádico en el 16%. Se conocen cuatro genes causantes: ADAMTS10, FBN1, LTBP2 y ADAMTS17.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
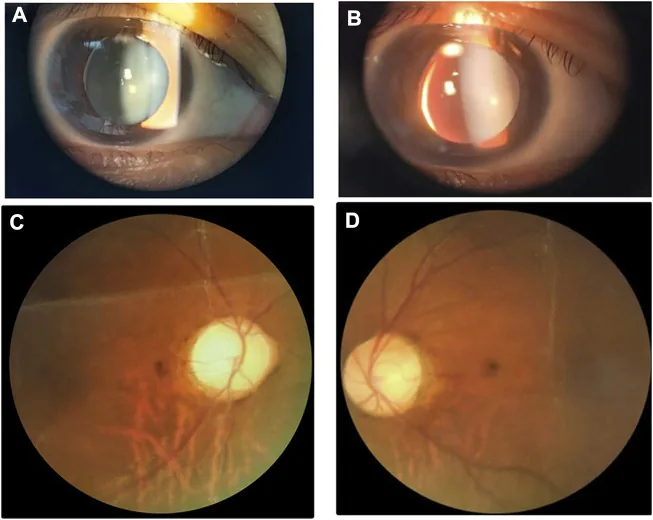
Imágenes de lámpara de hendidura y fondo de ojo que muestran un cristalino pequeño y grueso con subluxación inferonasal. La imagen es útil para reconocer las anomalías del cristalino que se superponen con el síndrome de Weill-Marchesani.
Los hallazgos oculares y sistémicos se resumen a continuación. La frecuencia de cada hallazgo se basa en las estadísticas de 128 casos de Faivre (2003). 2)
Hallazgos Oftalmológicos
Microesferofaquia: El diámetro ecuatorial es más pequeño de lo normal y el cristalino tiene forma esférica. Se ha medido un grosor del cristalino (LT) de 5.36 mm en un caso. Presente en el 84% de los casos. 1)
Luxación del cristalino: Presente en el 73% de los casos, con frecuencia hacia abajo. Puede acompañarse de iridodonesis.
Miopía alta: Presente en el 94% de los casos. Se han reportado casos que alcanzan OD -19.00 DS y OS -19.50 DS. 1)
Glaucoma secundario: Presente en el 80% de los casos. Principalmente glaucoma de ángulo cerrado por bloqueo pupilar. 2)
Cámara anterior poco profunda: Presenta una profundidad de cámara anterior de aproximadamente 2 CT (grosor corneal). 1)
Cuando el cristalino esferofáquico se desplaza hacia adelante, se produce un bloqueo pupilar, lo que desencadena glaucoma de ángulo cerrado. 2) Se informa que el 80% de los pacientes con síndrome de Weill-Marchesani desarrollan glaucoma. Los mióticos están contraindicados porque empeoran este mecanismo (ver sección “Tratamiento estándar” para más detalles).
La causa del síndrome de Weill-Marchesani son mutaciones en genes que codifican componentes de la matriz extracelular (MEC).
FBN1 (fibrilina-1): Herencia autosómica dominante. Codifica un componente principal de las microfibrillas y participa en la estructura de la zónula de Zinn.
ADAMTS10 y ADAMTS17: Herencia autosómica recesiva. Se sabe que están estrechamente relacionados con la fibrilina-1; su deficiencia provoca síntomas similares al síndrome de Weill-Marchesani.
LTBP2 (Proteína de unión a TGF-β latente 2): Herencia autosómica recesiva. Participa en la producción de matriz extracelular y está presente en tejidos elásticos y la zónula ciliar. 1)
Ejemplos de mutaciones específicas incluyen las mutaciones heterocigotas compuestas c.3672delC y c.3542delT en el gen LTBP2 (no registradas en la base de datos de Asia Oriental de GnomAD). 1) También se ha reportado una mutación homocigota c.2050C>T p(Arg684*) en ADAMTS10. 2)
El matrimonio consanguíneo es un factor de riesgo, y un informe de Arabia Saudita encontró que el 57% de los pacientes tenían antecedentes de matrimonio consanguíneo. 2)
El diagnóstico del síndrome de Weill-Marchesani se realiza clínicamente mediante la combinación de hallazgos oftalmológicos y sistémicos. La prueba genética es útil para la confirmación.
El análisis de exoma y la secuenciación de próxima generación (NGS) identifican variantes candidatas, que se confirman mediante secuenciación Sanger. 1)
Q¿Cuál es la diferencia entre el síndrome de Marfan y el síndrome de Weill-Marchesani?
A
El síndrome de Weill-Marchesani se presenta con baja estatura, braquidactilia y luxación del cristalino hacia abajo, mientras que el síndrome de Marfan se presenta con alta estatura, aracnodactilia y luxación del cristalino hacia arriba y afuera. El contraste en la constitución física ha llevado a denominarlo “síndrome de Marfan invertido”. La homocistinuria también causa luxación del cristalino (a menudo hacia adentro y abajo), pero la discapacidad intelectual y la trombosis ayudan en el diagnóstico diferencial. El síndrome de Weill-Marchesani se asocia con esferofaquia, por lo que es más propenso a complicarse con glaucoma en comparación con otros síndromes con luxación del cristalino.
La estrategia básica de tratamiento es una intervención escalonada según el estadio de la enfermedad. En casos leves, se recomienda observación; si el glaucoma o las complicaciones del cristalino progresan, se considera cirugía. Se recomienda la cirugía antes de que el cristalino caiga en la cavidad vítrea.
Extracción del cristalino + vitrectomía anterior + inserción de lente intraocular (LIO) fijada a la esclera: Se ha reportado una mejora de la presión intraocular postoperatoria a 13 mmHg en el ojo derecho y 12 mmHg en el izquierdo. 2)
Extracción intracapsular del cristalino: Se realiza para microesferofaquia.
Fisioterapia: Manejo de la rigidez articular y braquidactilia.
Seguimiento cardíaco: Detección y manejo de defectos cardiovasculares (24%).
Terapia con hormona de crecimiento (GH): La evidencia no está establecida y se encuentra en fase experimental. Existe un informe de inicio experimental en un caso con un pico de GH de 7.89 ng/mL. 2)
Q¿Por qué están contraindicados los mióticos?
A
Los mióticos (agentes colinérgicos) contraen el músculo ciliar y relajan las zónulas. Como resultado, el cristalino microesferofáquico se desplaza más hacia adelante, favoreciendo el bloqueo pupilar y empeorando el ataque de glaucoma de ángulo cerrado. En el síndrome de Weill-Marchesani, el principio del tratamiento es usar ciclopléjicos (midriáticos) para retraer el cristalino.
6. Fisiopatología y mecanismo detallado de la enfermedad
La patología del síndrome de Weill-Marchesani se entiende principalmente como un debilitamiento de las zónulas y la cápsula del cristalino debido a anomalías en los componentes de la matriz extracelular.
Las mutaciones de FBN1 alteran la estructura de las microfibrillas, causando debilidad zonular y aumento de la movilidad del cristalino. ADAMTS10 y ADAMTS17 están estrechamente relacionados con la fibrilina-1, y su deficiencia también produce síntomas similares al síndrome de Weill-Marchesani.
LTBP2 participa en la estabilidad de las microfibrillas de la MEC. Las mutaciones debilitan las zónulas y la cápsula del cristalino, provocando microesferofaquia y luxación del cristalino. 1)
Normalmente, alrededor del quinto al sexto mes de gestación, el cristalino se vuelve temporalmente esférico. Luego cambia a una forma elíptica debido al desarrollo mesodérmico normal, pero si hay anomalías mesodérmicas, esta forma esférica se mantiene, dando lugar a microesferofaquia. 1)
Tipo AR (ADAMTS10/LTBP2)
Patrón de herencia: Autosómico recesivo
Genes principales: ADAMTS10, LTBP2, ADAMTS17
Gravedad de los hallazgos oculares: Tiende a ser alta (LTBP2: LT 5.36 mm, PIO 26–30 mmHg) 1)
Hallazgos sistémicos: Baja estatura y braquidactilia son prominentes
Lin et al. (2021) informaron de una niña de 5 años con nuevas mutaciones heterocigotas compuestas en el gen LTBP2 (c.3672delC, c.3542delT). 1) Estas mutaciones eran nuevas y no estaban registradas en la base de datos GnomAD de Asia Oriental. La aplicación al diagnóstico prenatal y al asesoramiento genético se indica como una dirección futura.
Al Motawa et al. (2021) reportaron una familia con una mutación homocigota ADAMTS10 c.2050C>T p(Arg684*) y enfatizaron la importancia del consejo genético y el diagnóstico genético preimplantacional. 2) Los padres del paciente eran consanguíneos (primos hermanos).
Evidencia de la terapia con hormona de crecimiento
La terapia con hormona de crecimiento para la baja estatura asociada al síndrome de Weill-Marchesani carece actualmente de evidencia establecida. En el caso reportado por Al Motawa et al., el pico de hormona de crecimiento fue de 7.89 ng/mL, que no fue bajo, pero se inició la terapia con hormona de crecimiento de forma experimental. 2) Se necesitan más estudios para establecer su eficacia y seguridad.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.