Sindrom Weill-Marchesani (WMS) adalah penyakit jaringan ikat herediter. Juga disebut sindrom Spherophakia-Brachymorphia atau Mesodermal dysmorphodystrophy. Nama lain termasuk sindrom Marchesani dan sindrom Marfan terbalik. Dilaporkan oleh Weill dan Marchesani lebih dari 80 tahun yang lalu. 2)
Prevalensi diperkirakan 1 per 100.000 orang. 1) Pola pewarisan dapat berupa autosomal resesif (AR) atau autosomal dominan (AD), dengan 45% AR, 39% AD, dan 16% sporadis. 1)
Berdasarkan gen penyebab, sindrom ini diklasifikasikan menjadi 4 subtipe.
WMS1: ADAMTS10 (autosomal resesif) 2)
WMS2: FBN1 (autosomal dominan)
WMS3: LTBP2 (autosomal resesif) 1)
WMS4: ADAMTS17 (autosomal resesif)
Ciri khasnya adalah bentuk tubuh yang kontras dengan sindrom Marfan, sehingga disebut juga “Inverted Marfan syndrome”. Jika sindrom Marfan menunjukkan tinggi badan berlebih, jari-jari panjang seperti laba-laba, dan dislokasi lensa ke atas, maka sindrom Weill-Marchesani menunjukkan perawakan pendek, jari-jari pendek, dan dislokasi lensa ke bawah.
QSeberapa langka sindrom Weill-Marchesani?
A
Penyakit langka ini diperkirakan memiliki prevalensi 1 kasus per 100.000 orang. 1) Pola pewarisan terdiri dari autosomal resesif 45%, autosomal dominan 39%, dan sporadis 16%. Empat gen penyebab yang diketahui adalah ADAMTS10, FBN1, LTBP2, dan ADAMTS17.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
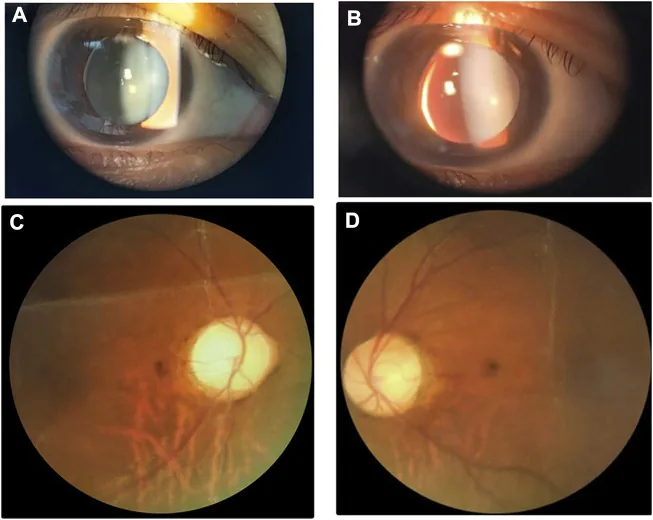
Gambar slit-lamp dan fundus menunjukkan lensa kecil dan tebal dengan subluksasi inferonasal. Gambar ini berguna untuk mengenali kelainan lensa yang tumpang tindih dengan sindrom Weill-Marchesani.
Temuan okular dan sistemik dirangkum di bawah ini. Frekuensi setiap temuan berdasarkan statistik 128 kasus oleh Faivre (2003). 2)
Temuan Okular
Lensa mikrosferofakia: Diameter ekuator lebih kecil dari normal dan berbentuk bulat. Terdapat contoh pengukuran LT (ketebalan lensa) 5,36 mm. Ditemukan pada 84% kasus. 1)
Dislokasi lensa: Ditemukan pada 73% kasus, sering ke arah inferior. Dapat disertai iridodonesis.
Miopia tinggi: Ditemukan pada 94% kasus. Terdapat laporan kasus mencapai OD -19,00 DS dan OS -19,50 DS. 1)
Glaukoma sekunder: ditemukan pada 80% kasus. Terutama glaukoma sudut tertutup akibat blokade pupil. 2)
Ketika lensa mikrosferofakia bergerak ke anterior, terjadi blokade pupil dan menyebabkan glaukoma sudut tertutup. 2) Sekitar 80% pasien dengan sindrom Weill-Marchesani mengalami glaukoma. Obat miotik dikontraindikasikan karena memperburuk mekanisme ini (lihat bagian “Terapi Standar” untuk detail).
Penyebab sindrom Weill-Marchesani adalah mutasi pada gen yang mengkode komponen matriks ekstraseluler (ECM).
FBN1 (fibrillin-1): Pewarisan autosomal dominan. Mengkode komponen utama mikrofibril, berperan dalam struktur zonula Zinn.
ADAMTS10·ADAMTS17: Pewarisan autosomal resesif. Diketahui terkait erat dengan fibrillin-1, defek menyebabkan gejala mirip sindrom Weill-Marchesani.
LTBP2 (Latent TGF-β Binding Protein 2): Pewarisan autosomal resesif. Berperan dalam produksi matriks ekstraseluler, terdapat pada jaringan elastis dan zonula siliaris. 1)
Contoh mutasi spesifik yang dilaporkan adalah mutasi heterozigot majemuk c.3672delC dan c.3542delT pada gen LTBP2 (tidak terdaftar di database GnomAD Asia Timur). 1) Juga dilaporkan mutasi homozigot ADAMTS10 c.2050C>T p(Arg684*). 2)
Perkawinan sedarah merupakan faktor risiko, dengan 57% pasien memiliki riwayat perkawinan sedarah dalam laporan dari Arab Saudi. 2)
Diagnosis sindrom Weil-Marchesani dilakukan secara klinis berdasarkan kombinasi temuan oftalmologis dan sistemik. Tes genetik berguna untuk konfirmasi.
QApa perbedaan antara sindrom Marfan dan sindrom Weill-Marchesani?
A
Sindrom Weill-Marchesani menunjukkan perawakan pendek, brakidaktili, dan dislokasi lensa ke bawah, sedangkan sindrom Marfan menunjukkan perawakan tinggi, arachnodaktili, dan dislokasi lensa ke atas dan ke luar. Karena kontras bentuk tubuh, sindrom ini juga disebut “Inverted Marfan syndrome”. Homosistinuria juga menunjukkan dislokasi lensa (sering ke bawah dan ke dalam), namun gangguan intelektual dan trombosis membantu dalam diagnosis banding. Sindrom Weill-Marchesani disertai dengan lensa sferofakia, sehingga lebih rentan terhadap glaukoma dibandingkan sindrom dislokasi lensa lainnya.
Prinsip dasar pengobatan adalah intervensi bertahap sesuai dengan perkembangan kondisi. Pada kasus ringan, dilakukan observasi; jika glaukoma atau komplikasi lensa berkembang, pertimbangkan operasi. Disarankan untuk melakukan operasi sebelum lensa jatuh ke dalam vitreus.
Fisioterapi: Penanganan kekakuan sendi dan brakidaktili.
Pemantauan jantung: Skrining dan penanganan defek kardiovaskular (24%).
Terapi hormon pertumbuhan (GH): Bukti belum mapan dan masih bersifat eksperimental. Terapi ini pernah dilaporkan dimulai secara eksperimental pada kasus dengan puncak GH 7,89 ng/mL. 2)
QMengapa obat miotik dikontraindikasikan?
A
Obat miotik (kolinergik) mengontraksikan otot siliaris dan merelaksasi zonula Zinn. Akibatnya, lensa yang berbentuk bola kecil dapat bergerak lebih ke depan, memperburuk blok pupil dan meningkatkan risiko serangan glaukoma sudut tertutup. Pada sindrom Weil-Marchesani, prinsip pengobatan adalah menggunakan obat sikloplegik (midriatik) untuk mendorong lensa ke belakang.
Mutasi FBN1 mengganggu struktur mikrofibril, menyebabkan kelemahan zonula Zinn dan peningkatan mobilitas lensa. ADAMTS10 dan ADAMTS17 terkait erat dengan fibrillin-1, dan defisiensi keduanya juga menimbulkan gejala mirip sindrom Weil-Marchesani.
LTBP2 berperan dalam stabilitas mikrofibril ECM. Mutasi menyebabkan kelemahan zonula Zinn dan kapsul lensa, mengakibatkan lensa berbentuk bola kecil dan ektopia lensa. 1)
Normalnya, pada usia kehamilan 5–6 bulan, lensa mata berbentuk sferis sementara. Kemudian, karena perkembangan mesoderm yang normal, lensa berubah menjadi bentuk elips. Namun, kelainan pada mesoderm dapat menyebabkan bentuk sferis ini bertahan, sehingga terbentuk lensa sferofakia mikrofakia. 1)
Tipe AR (ADAMTS10/LTBP2)
Pola pewarisan: Autosomal resesif
Gen utama: ADAMTS10, LTBP2, ADAMTS17
Tingkat keparahan temuan mata: Cenderung tinggi (LTBP2: LT 5,36 mm, tekanan intraokular 26–30 mmHg) 1)
Temuan sistemik: Perawakan pendek dan brakidaktili yang menonjol
Tipe AD (FBN1)
Pola pewarisan: Autosomal dominan
Gen utama: FBN1 (fibrillin-1)
Karakteristik: Mutasi gen yang sama dengan sindrom Marfan
Temuan sistemik: Disertai perawakan pendek, brakidaktili, dan kekakuan sendi
Selain itu, telah diajukan hipotesis bahwa kelainan distribusi aktin berperan dalam patogenesis sindrom Weill-Marchesani. 2)
7. Penelitian Terkini dan Prospek ke Depan (Laporan Tahap Penelitian)
Lin dkk. (2021) melaporkan seorang anak perempuan berusia 5 tahun dengan mutasi heterozigot majemuk baru pada gen LTBP2 (c.3672delC · c.3542delT). 1) Mutasi ini merupakan mutasi baru yang belum terdaftar di basis data GnomAD Asia Timur. Penerapan untuk diagnosis prenatal dan konseling genetik diindikasikan sebagai arah masa depan.
Al Motawa dkk. (2021) melaporkan sebuah keluarga dengan mutasi homozigot ADAMTS10 c.2050C>T p(Arg684*), dan menekankan pentingnya konseling genetik serta diagnosis genetik praimplantasi. 2) Orang tua pasien adalah pernikahan kerabat (pernikahan sepupu).
Terapi hormon pertumbuhan untuk perawakan pendek pada sindrom Weil-Marchesani saat ini belum memiliki bukti yang mapan. Pada kasus yang dilaporkan oleh Al Motawa dkk., puncak hormon pertumbuhan adalah 7,89 ng/mL, yang tidak rendah, namun terapi hormon pertumbuhan dimulai secara eksperimental. 2) Penelitian lebih lanjut diperlukan untuk menetapkan efektivitas dan keamanannya.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.