سندرم ویل-مارکزانی (Weill-Marchesani Syndrome; WMS) یک بیماری ارثی بافت همبند است. همچنین به عنوان سندرم اسفروفاکی-براکیمورفی یا دیسمورفودیستروفی مزانشیمی شناخته میشود. نامهای دیگر آن سندرم مارکزانی و سندرم مارفان معکوس است. بیش از ۸۰ سال پیش توسط ویل و مارکزانی گزارش شد. 2)
شیوع آن حدود ۱ در ۱۰۰٬۰۰۰ نفر تخمین زده میشود. 1) الگوی توارث میتواند اتوزومال مغلوب (AR) یا اتوزومال غالب (AD) باشد؛ به ترتیب ۴۵٪ اتوزومال مغلوب، ۳۹٪ اتوزومال غالب و ۱۶٪ موارد پراکنده گزارش شده است. 1)
بر اساس ژن عامل، به چهار زیرگروه تقسیم میشود.
WMS1: ADAMTS10 (اتوزومال مغلوب) 2)
WMS2: FBN1 (اتوزومال غالب)
WMS3: LTBP2 (اتوزومال مغلوب) 1)
WMS4: ADAMTS17 (اتوزومال مغلوب)
این سندرم با داشتن تیپ بدنی متضاد با سندرم مارفان مشخص میشود و «سندرم مارفان معکوس» نیز نامیده میشود. در حالی که سندرم مارفان با قد بلند، انگشتان عنکبوتی و جابجایی عدسی به سمت بالا همراه است، سندرم ویل-مارکزانی با قد کوتاه، انگشتان کوتاه و جابجایی عدسی به سمت پایین مشخص میشود.
Qسندرم ویل-مارکزانی چقدر نادر است؟
A
این یک بیماری نادر با شیوع تخمینی ۱ در ۱۰۰٬۰۰۰ نفر است. 1) الگوی توارث در ۴۵٪ موارد اتوزومال مغلوب، در ۳۹٪ اتوزومال غالب و در ۱۶٪ موارد پراکنده است. چهار ژن شناخته شده شامل ADAMTS10، FBN1، LTBP2 و ADAMTS17 در ایجاد این سندرم نقش دارند.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
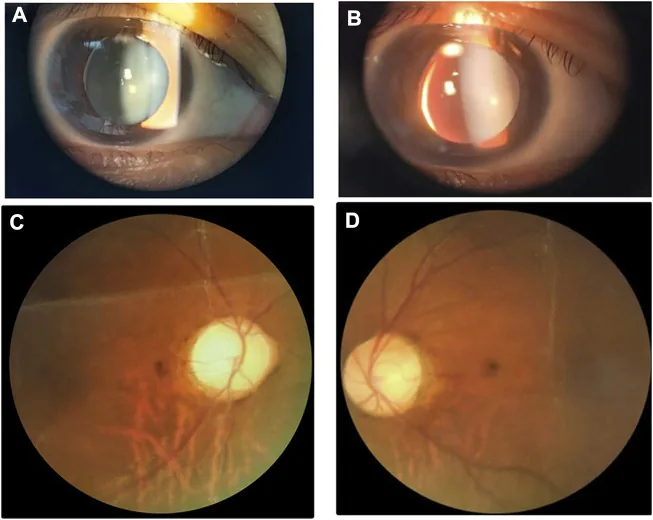
تصاویر اسلیت لمپ و فوندوس که یک عدسی کوچک و ضخیم با سابلوکساسیون به سمت پایین-داخلی را نشان میدهد. این تصویر برای شناسایی ناهنجاریهای عدسی که با سندرم ویل-مارکزانی همپوشانی دارند مفید است.
یافتههای چشمی و سیستمیک در زیر خلاصه شده است. فراوانی هر یافته بر اساس آمار 128 موردی Faivre (2003) است. 2)
یافتههای چشمی
میکروسفری (عدسی کروی کوچک): قطر استوایی کوچکتر از حد طبیعی و به شکل کروی است. نمونهای با ضخامت عدسی (LT) 5.36 میلیمتر اندازهگیری شده است. در 84% موارد دیده میشود. 1)
جابجایی عدسی: در 73% موارد دیده میشود و اغلب به سمت پایین است. ممکن است با لرزش عنبیه همراه باشد.
نزدیکبینی شدید: در 94% موارد دیده میشود. مواردی با OD -19.00 دیوپتر و OS -19.50 دیوپتر گزارش شده است. 1)
گلوکوم ثانویه: در 80% موارد دیده میشود. عمدتاً گلوکوم زاویه بسته ناشی از بلوک مردمکی است. 2)
اتاق قدامی کم عمق: حدود 2 CT (ضخامت قرنیه) اتاق قدامی کم عمق را نشان میدهد. 1)
هنگامی که عدسی کروی کوچک به سمت جلو حرکت میکند، بلوک مردمک ایجاد شده و گلوکوم زاویه بسته رخ میدهد.2) گفته میشود که ۸۰٪ از بیماران مبتلا به سندرم ویل-مارکزانی دچار گلوکوم میشوند. داروهای میوتیک این مکانیسم را تشدید میکنند، بنابراین منع مصرف دارند (برای جزئیات به بخش «روشهای درمان استاندارد» مراجعه کنید).
علت سندرم ویل-مارکزانی جهش در ژنهای کدکننده اجزای ماتریکس خارج سلولی (ECM) است.
FBN1 (فیبریلین-1): وراثت اتوزومال غالب. کدکننده جزء اصلی میکروفیبریلها و دخیل در ساختار زونول Zinn.
ADAMTS10 و ADAMTS17: وراثت اتوزومال مغلوب. ارتباط نزدیک با فیبریلین-1 شناخته شده است و کمبود آن باعث علائم شبیه سندرم ویل-مارکزانی میشود.
LTBP2 (پروتئین اتصالدهنده TGF-β نهفته 2): وراثت اتوزومال مغلوب. در تولید ماتریکس خارج سلولی نقش دارد و در بافت الاستیک و زونول مژگانی وجود دارد. 1)
نمونههای خاص جهش شامل جهشهای هتروزیگوت مرکب c.3672delC و c.3542delT در ژن LTBP2 (ثبتنشده در پایگاه داده GnomAD شرق آسیا) گزارش شده است. 1) همچنین جهش هموزیگوت c.2050C>T p(Arg684*) در ADAMTS10 گزارش شده است. 2)
ازدواج فامیلی یک عامل خطر است و در گزارش عربستان سعودی، 57% بیماران سابقه ازدواج فامیلی داشتند. 2)
Qتفاوت بین سندرم مارفان و سندرم ویل-مارکزانی چیست؟
A
سندرم ویل-مارکزانی با قد کوتاه، انگشتان کوتاه و جابجایی عدسی به سمت پایین مشخص میشود، در حالی که سندرم مارفان با قد بلند، انگشتان عنکبوتی و جابجایی عدسی به سمت بالا و خارج همراه است. به دلیل تضاد در تیپ بدنی، گاهی «سندرم مارفان معکوس» نامیده میشود. هوموسیستینوریا نیز باعث جابجایی عدسی (اغلب به سمت داخل و پایین) میشود، اما وجود ناتوانی ذهنی و ترومبوز به تشخیص افتراقی کمک میکند. همچنین سندرم ویل-مارکزانی با عدسی کروی همراه است، بنابراین نسبت به سایر سندرمهای همراه با جابجایی عدسی، بیشتر مستعد ابتلا به گلوکوم است.
اصل اساسی درمان، مداخله مرحلهای بر اساس پیشرفت بیماری است. در موارد خفیف، پیگیری کافی است و در صورت پیشرفت گلوکوم یا عوارض عدسی، جراحی در نظر گرفته میشود. توصیه میشود جراحی قبل از افتادن عدسی به داخل زجاجیه انجام شود.
استخراج عدسی + ویترکتومی قدامی + کاشت لنز داخل چشمی (IOL) با تثبیت اسکلرال: گزارش شده است که فشار داخل چشم پس از عمل به ۱۳ میلیمتر جیوه در چشم راست و ۱۲ میلیمتر جیوه در چشم چپ بهبود یافته است. 2)
استخراج عدسی داخل کپسولی: برای عدسیهای کروی کوچک انجام میشود.
ایریدکتومی محیطی و ایریدوتومی محیطی با لیزر (LPI): برای رفع بلوک مردمک استفاده میشود. 2)
ترابکولکتومی: در موارد کنترل ضعیف گلوکوم انجام میشود و گزارش شده است که نیمی از موارد نیاز به جراحی دارند. 2)
پیگیری قلبی: غربالگری و مدیریت نقایص قلبی عروقی (۲۴٪).
درمان با هورمون رشد (GH): شواهد قطعی نیست و در مرحله آزمایشی است. در یک مورد با پیک GH 7.89 ng/mL، درمان آزمایشی شروع شده است. 2)
Qچرا داروهای میوتیک (انقباضدهنده مردمک) منع مصرف دارند؟
A
داروهای میوتیک (کولینرژیک) باعث انقباض عضله مژگانی و شل شدن زونولهای زین میشوند. در نتیجه، عدسی کروی کوچک بیشتر به سمت جلو حرکت کرده و بلوک مردمک تشدید میشود که خطر حمله گلوکوم زاویه بسته را افزایش میدهد. در سندرم ویل-مارکزانی، استفاده از داروهای سیکلوپلژیک (گشادکننده مردمک) برای عقب بردن عدسی، اصل درمان است.
جهش FBN1 ساختار میکروفیبریلها را مختل کرده و باعث ضعف زونولهای زین و افزایش تحرک عدسی میشود. ADAMTS10 و ADAMTS17 ارتباط نزدیکی با فیبریلین-۱ دارند و کمبود آنها نیز علائمی شبیه سندرم ویل-مارکزانی ایجاد میکند.
LTBP2 در پایداری میکروفیبریلهای ماتریکس خارج سلولی نقش دارد. جهش آن باعث ضعف زونولهای زین و کپسول عدسی شده و منجر به عدسی کروی کوچک و جابجایی عدسی میشود. 1)
به طور طبیعی در حدود ماههای ۵ تا ۶ جنینی، عدسی به طور موقت کروی میشود. سپس با رشد طبیعی مزودرم به شکل بیضی تغییر میکند، اما در صورت ناهنجاری مزودرم، این شکل کروی حفظ شده و عدسی کروی کوچک ایجاد میشود. 1)
نوع اتوزومال مغلوب (ADAMTS10/LTBP2)
الگوی وراثت: اتوزومال مغلوب
ژنهای اصلی: ADAMTS10، LTBP2، ADAMTS17
شدت یافتههای چشمی: تمایل به بالا (LTBP2: LT 5.36 میلیمتر، فشار چشم ۲۶-۳۰ میلیمتر جیوه) 1)
یافتههای سیستمیک: کوتاهی قد و براکیداکتیلی قابل توجه
لین و همکاران (2021) یک دختر 5 ساله با جهشهای هتروزیگوت مرکب جدید در ژن LTBP2 (c.3672delC و c.3542delT) گزارش کردند. 1) این جهشها جدید بوده و در پایگاه داده GnomAD شرق آسیا ثبت نشده بودند. کاربرد در تشخیص پیش از تولد و مشاوره ژنتیک به عنوان جهتگیری آینده نشان داده شده است.
ال موتاوا و همکاران (2021) خانوادهای با جهش هموزیگوت ADAMTS10 c.2050C>T p(Arg684*) را گزارش کردند و بر اهمیت مشاوره ژنتیک و تشخیص ژنتیکی پیش از لانهگزینی تأکید کردند. 2) والدین بیمار ازدواج فامیلی (ازدواج با خویشاوند) داشتند.
درمان با هورمون رشد برای کوتاهی قد مرتبط با سندرم ویل-مارکزانی در حال حاضر شواهد قطعی ندارد. در مورد گزارش شده توسط ال موتاوا و همکاران، اوج هورمون رشد 7.89 ng/mL بود که پایین نبود، اما درمان با هورمون رشد به صورت آزمایشی آغاز شد. 2) اثربخشی و ایمنی نیاز به بررسی بیشتر در آینده دارد.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.