Синдром Вейля-Маркезани (Weill-Marchesani Syndrome; WMS) — это наследственное заболевание соединительной ткани. Также называется сферофакия-брахиморфия синдром или мезодермальная дисморфодистрофия. Другие названия: синдром Маркезани, инвертированный синдром Марфана. Был описан Вейлем и Маркезани более 80 лет назад. 2)
Распространенность оценивается как 1 случай на 100 000 человек. 1) Тип наследования может быть как аутосомно-рецессивным (АР), так и аутосомно-доминантным (АД): АР — 45%, АД — 39%, спорадические случаи — 16%. 1)
В зависимости от гена-причины выделяют 4 подтипа.
WMS1: ADAMTS10 (аутосомно-рецессивный) 2)
WMS2: FBN1 (аутосомно-доминантный)
WMS3: LTBP2 (аутосомно-рецессивный) 1)
WMS4: ADAMTS17 (аутосомно-рецессивный)
Характерной особенностью является телосложение, противоположное синдрому Марфана, поэтому его также называют «инвертированным синдромом Марфана». Если синдром Марфана проявляется высоким ростом, арахнодактилией и смещением хрусталика вверх, то синдром Вайля-Маркезани — низким ростом, брахидактилией и смещением хрусталика вниз.
QНасколько редко встречается синдром Вайля-Маркезани?
A
Это редкое заболевание с предполагаемой распространённостью 1 случай на 100 000 человек. 1) Тип наследования: аутосомно-рецессивный в 45% случаев, аутосомно-доминантный в 39%, спорадический в 16%. Известны четыре гена-кандидата: ADAMTS10, FBN1, LTBP2 и ADAMTS17.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
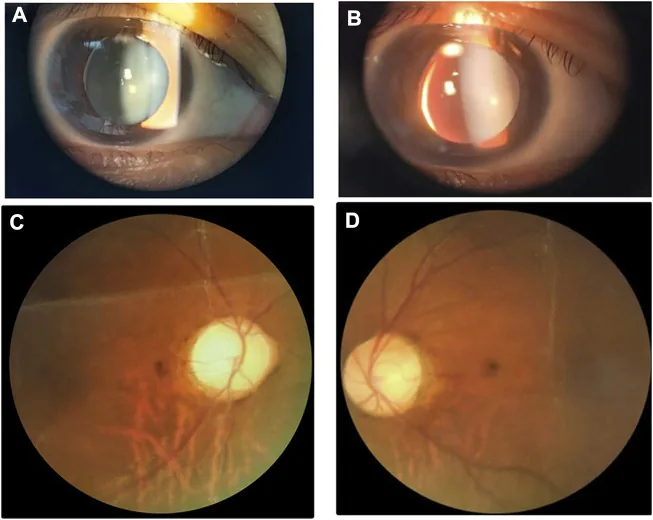
Изображения щелевой лампы и глазного дна, показывающие маленький толстый хрусталик с нижне-носовым подвывихом. Изображение полезно для распознавания аномалий хрусталика, характерных для синдрома Вайля-Маркезани.
Ниже приведены глазные и системные проявления. Частота каждого признака основана на статистике 128 случаев Faivre (2003). 2)
Офтальмологические признаки
Микрофакия (маленький хрусталик): экваториальный диаметр меньше обычного, хрусталик имеет шаровидную форму. Зарегистрирован случай с толщиной хрусталика (LT) 5,36 мм. Наблюдается в 84% случаев. 1)
Эктопия хрусталика: встречается в 73% случаев, чаще смещение вниз. Может сопровождаться иридодонезом.
Высокая миопия: наблюдается в 94% случаев. Сообщалось о случаях с OD -19,00 дптр и OS -19,50 дптр. 1)
Вторичная глаукома: наблюдается в 80% случаев. Преимущественно закрытоугольная глаукома вследствие зрачкового блока. 2)
Разжижение стекловидного тела и утолщение роговицы также описаны.
Системные проявления
Низкий рост: наблюдается в 98% случаев. Сообщается о росте 103 см (Z-оценка -5,4). 2)
Брахидактилия и клинодактилия: встречаются в 98% случаев. 2)
Тугоподвижность суставов: наблюдается в 62% случаев. 2)
Утолщение кожи, псевдоатлетическое телосложение: характерный внешний вид.
Аномалии лица: узкие глазные щели, длинные ресницы, широкое переносье, тонкая верхняя губа. 2)
Сердечно-сосудистые дефекты: встречаются у 24%. Включая открытый артериальный проток и др. 2)
Умственная отсталость: встречается у 13%. 2)
Также сообщается о нарушениях слуха.
Ниже приведена частота основных признаков.
Признак
Частота (%)
Низкий рост
98
Брахидактилия
98
Миопия
94
Микрофакия
84
Вторичная глаукома
80
Смещение хрусталика
73
Тугоподвижность суставов
62
Сердечно-сосудистые дефекты
24
Катаракта
23
Умственная отсталость
13
QПочему возникает глаукома?
A
Когда сферофакия смещается вперед, возникает зрачковый блок, что приводит к закрытоугольной глаукоме. 2) Считается, что глаукома развивается у 80% пациентов с синдромом Вейля-Маркезани. Миотики усугубляют этот механизм, поэтому они противопоказаны (подробнее см. в разделе «Стандартные методы лечения»).
Причиной синдрома Вейля-Маркезани являются мутации в генах, кодирующих компоненты внеклеточного матрикса (ВКМ).
FBN1 (фибриллин-1): аутосомно-доминантное наследование. Кодирует основной компонент микрофибрилл, участвует в структуре цинновой связки.
ADAMTS10 и ADAMTS17: аутосомно-рецессивное наследование. Известна тесная связь с фибриллином-1, их дефицит вызывает симптомы, подобные синдрому Вейля-Маркезани.
LTBP2 (латентный TGF-β-связывающий белок 2): аутосомно-рецессивное наследование. Участвует в продукции внеклеточного матрикса, присутствует в эластических тканях и цинновой связке. 1)
В качестве конкретных примеров мутаций сообщается о компаунд-гетерозиготных мутациях c.3672delC и c.3542delT в гене LTBP2 (не зарегистрированы в базе данных GnomAD East Asia). 1) Также сообщается о гомозиготной мутации ADAMTS10 c.2050C>T p(Arg684*). 2)
Родственные браки являются фактором риска: по данным из Саудовской Аравии, у 57% пациентов были родственные браки в анамнезе. 2)
Диагноз синдрома Вейля-Маркезани ставится клинически на основе сочетания офтальмологических и общих симптомов. Для подтверждения полезна генетическая диагностика.
Исследования, используемые для клинической диагностики
QВ чем разница между синдромом Марфана и синдромом Вейля-Маркезани?
A
Синдром Вейля-Маркезани характеризуется низким ростом, брахидактилией и смещением хрусталика вниз, тогда как синдром Марфана — высоким ростом, арахнодактилией и смещением хрусталика вверх и наружу. Из-за контрастного телосложения его также называют «инвертированным синдромом Марфана». Гомоцистинурия также вызывает смещение хрусталика (чаще внутрь и вниз), но наличие умственной отсталости и тромбозов помогает в дифференциальной диагностике. Поскольку синдром Вейля-Маркезани сопровождается сферофакией, он чаще осложняется глаукомой по сравнению с другими синдромами, вызывающими смещение хрусталика.
Основной принцип лечения — поэтапное вмешательство в зависимости от степени прогрессирования заболевания. При легкой форме рекомендуется наблюдение, при прогрессировании глаукомы или осложнений со стороны хрусталика рассматривается хирургическое вмешательство. Рекомендуется проводить операцию до выпадения хрусталика в стекловидное тело.
Экстракция хрусталика + передняя витрэктомия + интраокулярная линза (ИОЛ) с фиксацией к склере: имеются сообщения об улучшении послеоперационного внутриглазного давления до 13 мм рт. ст. на правом глазу и 12 мм рт. ст. на левом. 2)
Интракапсулярная экстракция хрусталика: выполняется при микросферофакии.
Периферическая иридэктомия / лазерная периферическая иридотомия (ЛПИ): используется для устранения зрачкового блока. 2)
Трабекулэктомия: выполняется при плохом контроле глаукомы; по сообщениям, операция требуется в половине случаев. 2)
Физиотерапия: лечение тугоподвижности суставов и брахидактилии.
Кардиологическое наблюдение: скрининг и лечение сердечно-сосудистых дефектов (24%).
Терапия гормоном роста (ГР): доказательная база не установлена, находится на экспериментальной стадии. Имеется сообщение о начале терапии в пробном порядке у пациента с пиком ГР 7,89 нг/мл. 2)
QПочему миотики противопоказаны?
A
Миотики (холинергические средства) вызывают сокращение цилиарной мышцы и расслабление цинновой связки. В результате малый сферический хрусталик смещается еще больше вперед, что способствует блоку зрачка и может ухудшить приступ закрытоугольной глаукомы. При синдроме Вейля-Маркезани принципом лечения является использование циклоплегиков (мидриатиков) для смещения хрусталика назад.
Патогенез синдрома Вейля-Маркезани в основном связан с ослаблением цинновой связки и капсулы хрусталика из-за аномалий компонентов внеклеточного матрикса.
Мутации FBN1 нарушают структуру микрофибрилл, вызывая ослабление цинновой связки и повышение подвижности хрусталика. ADAMTS10 и ADAMTS17 тесно связаны с фибриллином-1, и их дефицит также приводит к симптомам, подобным синдрому Вейля-Маркезани.
LTBP2 участвует в стабильности микрофибрилл внеклеточного матрикса. Мутации вызывают ослабление цинновой связки и капсулы хрусталика, что приводит к малому сферическому хрусталику и его смещению. 1)
В норме на 5–6 месяце эмбрионального развития хрусталик временно становится сферическим. Впоследствии, благодаря нормальному развитию мезодермы, он приобретает эллиптическую форму, однако при аномалии мезодермы эта сферическая форма сохраняется, что приводит к микросферофакии. 1)
Аутосомно-рецессивный тип (ADAMTS10/LTBP2)
Тип наследования: аутосомно-рецессивный
Основные гены: ADAMTS10, LTBP2, ADAMTS17
Тяжесть глазных проявлений: высокая (LTBP2: толщина хрусталика 5,36 мм, внутриглазное давление 26–30 мм рт. ст.) 1)
Системные проявления: выраженные низкорослость и брахидактилия
Аутосомно-доминантный тип (FBN1)
Тип наследования: аутосомно-доминантный
Основной ген: FBN1 (фибриллин-1)
Особенности: мутация того же гена, что и при синдроме Марфана
Lin и соавт. (2021) сообщили о 5-летней девочке с новыми компаунд-гетерозиготными мутациями в гене LTBP2 (c.3672delC и c.3542delT). 1) Эти мутации являются новыми и не зарегистрированы в базе данных GnomAD для Восточной Азии. В качестве будущего направления указано применение в пренатальной диагностике и генетическом консультировании.
Генетическое консультирование в семье с мутацией ADAMTS10
Al Motawa и соавт. (2021) сообщили о семье с гомозиготной мутацией ADAMTS10 c.2050C>T p(Arg684*) и подчеркнули важность генетического консультирования и преимплантационной генетической диагностики. 2) Родители пациента состояли в кровном родстве (двоюродный брак).
Терапия гормоном роста при низкорослости, связанной с синдромом Вейля-Маркезани, в настоящее время не имеет установленных доказательств. В случае, описанном Al Motawa и соавт., пик гормона роста составил 7,89 нг/мл, что не было низким, однако терапия гормоном роста была начата в порядке пробы. 2) Для установления эффективности и безопасности необходимы дальнейшие исследования.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.