Das Weill-Marchesani-Syndrom (WMS) ist eine erbliche Bindegewebserkrankung. Es wird auch als Spherophakia-Brachymorphia-Syndrom oder Mesodermale Dysmorphodystrophie bezeichnet. Weitere Namen sind Marchesani-Syndrom und Invertiertes Marfan-Syndrom. Es wurde vor über 80 Jahren von Weill und Marchesani beschrieben. 2)
Die Prävalenz wird auf 1 Fall pro 100.000 Personen geschätzt. 1) Der Erbgang kann sowohl autosomal-rezessiv (AR) als auch autosomal-dominant (AD) sein, wobei autosomal-rezessiv 45 %, autosomal-dominant 39 % und sporadisch 16 % betragen. 1)
Je nach verursachendem Gen werden vier Subtypen unterschieden.
WMS1: ADAMTS10 (autosomal-rezessiv) 2)
WMS2: FBN1 (autosomal-dominant)
WMS3: LTBP2 (autosomal-rezessiv) 1)
WMS4: ADAMTS17 (autosomal-rezessiv)
Es zeichnet sich durch einen gegensätzlichen Körperbau zum Marfan-Syndrom aus und wird auch als „Inverted Marfan-Syndrom“ bezeichnet. Während das Marfan-Syndrom durch Hochwuchs, Spinnenfingrigkeit und eine nach oben verlagerte Linse gekennzeichnet ist, zeigt das Weill-Marchesani-Syndrom Kleinwuchs, Brachydaktylie und eine nach unten verlagerte Linse.
QWie selten ist das Weill-Marchesani-Syndrom?
A
Es handelt sich um eine seltene Erkrankung mit einer geschätzten Prävalenz von 1 Fall pro 100.000 Personen. 1) Der Erbgang ist autosomal-rezessiv (45 %), autosomal-dominant (39 %) oder sporadisch (16 %). Vier ursächliche Gene sind bekannt: ADAMTS10, FBN1, LTBP2 und ADAMTS17.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
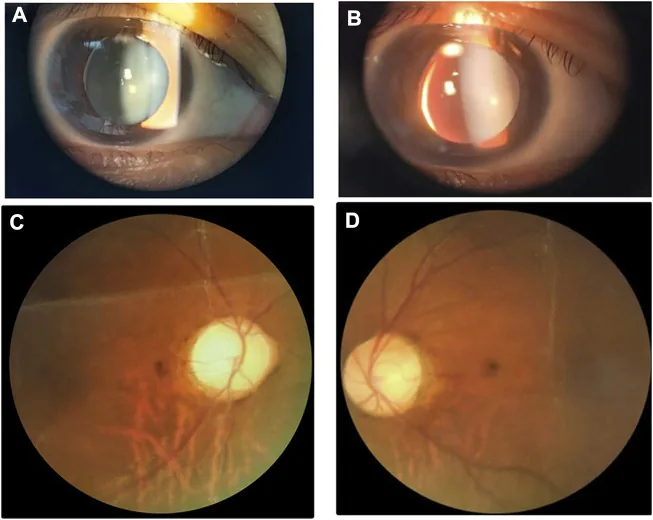
Spaltlampen- und Fundusaufnahmen zeigen eine kleine, dicke Linse mit inferonasaler Subluxation. Das Bild ist hilfreich, um die Linsenveränderungen zu erkennen, die mit dem Weill-Marchesani-Syndrom einhergehen.
Augenbefunde und Allgemeinbefunde sind im Folgenden zusammengefasst. Die Häufigkeit der einzelnen Befunde basiert auf der Statistik von 128 Fällen nach Faivre (2003). 2)
Augenbefunde
Mikrosphärophakie: Der Äquatordurchmesser ist kleiner als normal und die Linse kugelförmig. Es gibt Messbeispiele mit einer Linsendicke (LT) von 5,36 mm. Tritt bei 84% auf. 1)
Linsenluxation: tritt bei 73% auf, häufig nach unten verlagert. Kann von Iridodonesis begleitet sein.
Hohe Myopie: tritt bei 94% auf. Es gibt Berichte über Werte von OD -19,00 DS und OS -19,50 DS. 1)
Sekundärglaukom: Tritt bei 80% auf. Hauptsächlich Winkelblockglaukom durch Pupillarblock. 2)
Wenn die Mikrosphärophakie nach vorne wandert, kommt es zu einem Pupillarblock, der ein Winkelblockglaukom auslöst. 2) Bei 80% der Patienten mit Weill-Marchesani-Syndrom tritt ein Glaukom auf. Miotika sind kontraindiziert, da sie diesen Mechanismus verschlechtern (siehe Abschnitt „Standardtherapie“).
Die Ursache des Weill-Marchesani-Syndroms sind Mutationen in Genen, die für Komponenten der extrazellulären Matrix (ECM) kodieren.
FBN1 (Fibrillin-1): autosomal-dominante Vererbung. Kodiert die Hauptkomponente der Mikrofibrillen und ist an der Struktur der Zonula Zinnii beteiligt.
ADAMTS10/ADAMTS17: autosomal-rezessive Vererbung. Stehen in enger Verbindung mit Fibrillin-1; ein Defekt führt zu Symptomen ähnlich dem Weill-Marchesani-Syndrom.
LTBP2 (Latent TGF-β Binding Protein 2): autosomal-rezessive Vererbung. Beteiligt an der Produktion der extrazellulären Matrix, vorhanden im elastischen Gewebe und den Zonulafasern. 1)
Als spezifisches Mutationsbeispiel wurden compound-heterozygote Mutationen c.3672delC und c.3542delT im LTBP2-Gen (nicht in der GnomAD-Datenbank für Ostasien registriert) berichtet. 1) Auch eine homozygote Mutation ADAMTS10 c.2050C>T p(Arg684*) wurde beschrieben. 2)
Blutsverwandtschaft ist ein Risikofaktor; in einem Bericht aus Saudi-Arabien hatten 57% der Patienten eine konsanguine Ehe in der Vorgeschichte. 2)
Die Diagnose des Weil-Marchesani-Syndroms wird klinisch anhand einer Kombination von ophthalmologischen und systemischen Befunden gestellt. Zur Bestätigung ist ein Gentest hilfreich.
QWas ist der Unterschied zwischen Marfan-Syndrom und Weill-Marchesani-Syndrom?
A
Das Weill-Marchesani-Syndrom zeigt Kleinwuchs, Brachydaktylie und Linsendislokation nach unten, während das Marfan-Syndrom durch Hochwuchs, Arachnodaktylie und Linsenluxation nach oben-außen gekennzeichnet ist. Aufgrund des gegensätzlichen Körperbaus wird es auch als „invertiertes Marfan-Syndrom“ bezeichnet. Homocystinurie führt ebenfalls zu Linsenluxation (häufiger nach innen-unten), aber geistige Behinderung und Thrombosen helfen bei der Abgrenzung. Da das Weill-Marchesani-Syndrom mit einer Mikrophakie einhergeht, besteht im Vergleich zu anderen Syndromen mit Linsenluxation ein erhöhtes Risiko für Glaukom.
Das grundlegende Behandlungsprinzip ist ein stufenweises Vorgehen je nach Krankheitsfortschritt. Bei milden Fällen wird abgewartet, bei fortgeschrittenem Glaukom oder Linsenkomplikationen wird eine Operation in Betracht gezogen. Es wird empfohlen, die Operation durchzuführen, bevor die Linse in den Glaskörper fällt.
Linsenentfernung + vordere Vitrektomie + Sklerafixierte Intraokularlinse (IOL): Es gibt Berichte über eine Verbesserung des postoperativen Augeninnendrucks auf 13 mmHg rechts und 12 mmHg links. 2)
Intrakapsuläre Linsenentfernung: Wird bei Mikrosphärophakie durchgeführt.
Periphere Iridektomie / Laser-periphere Iridotomie (LPI): Zur Aufhebung eines Pupillarblocks. 2)
Trabekulektomie: Wird bei unzureichender Glaukomkontrolle durchgeführt; Berichten zufolge ist bei der Hälfte der Fälle eine Operation erforderlich. 2)
Bei Kindern wird zur Vorbeugung einer Amblyopie frühzeitig eine Refraktionskorrektur (Brille) durchgeführt und zum geeigneten Zeitpunkt zur Operation übergegangen.
Physiotherapie: Behandlung von Gelenksteifigkeit und Brachydaktylie.
Kardiologische Nachsorge: Screening und Management von kardiovaskulären Defekten (24%).
Wachstumshormontherapie (GH): Die Evidenz ist nicht etabliert und befindet sich im experimentellen Stadium. Es gibt Berichte über einen Versuchsbeginn bei einem Fall mit einem GH-Peak von 7,89 ng/mL. 2)
QWarum sind Miotika kontraindiziert?
A
Miotika (cholinergische Wirkstoffe) kontrahieren den Ziliarmuskel und entspannen die Zonulafasern. Dadurch kann die Mikrosphärophakie weiter nach vorne wandern, was einen Pupillarblock begünstigt und das Risiko eines akuten Winkelblockglaukoms erhöht. Beim Weil-Marchesani-Syndrom ist die Behandlung mit Zykloplegika (Mydriatika) zur Rückverlagerung der Linse das Prinzip.
6. Pathophysiologie und detaillierter Pathomechanismus
Die Pathophysiologie des Weil-Marchesani-Syndroms wird durch eine Schwächung der Zonulafasern und der Linsenkapsel aufgrund von Anomalien der extrazellulären Matrixbestandteile verstanden.
FBN1-Mutationen beeinträchtigen die Struktur der Mikrofibrillen, was zu einer Schwächung der Zonulafasern und einer erhöhten Linsenbeweglichkeit führt. ADAMTS10 und ADAMTS17 sind eng mit Fibrillin-1 assoziiert; deren Defekte verursachen ebenfalls Symptome ähnlich dem Weil-Marchesani-Syndrom.
LTBP2 ist an der Stabilität der Mikrofibrillen in der extrazellulären Matrix beteiligt. Mutationen führen zu einer Schwächung der Zonulafasern und der Linsenkapsel, was Mikrosphärophakie und Linsenluxation verursacht. 1)
Normalerweise wird die Linse im 5. bis 6. Schwangerschaftsmonat vorübergehend kugelförmig. Anschließend verändert sie sich durch normale Entwicklung des Mesoderms zu einer elliptischen Form. Bei einer Anomalie des Mesoderms bleibt diese Kugelform erhalten und es entsteht eine Mikrosphärophakie. 1)
AR-Typ (ADAMTS10/LTBP2)
Vererbungsmodus: autosomal-rezessiv
Hauptgene: ADAMTS10, LTBP2, ADAMTS17
Schweregrad der Augenmanifestationen: tendenziell hoch (LTBP2: LT 5,36 mm, Augeninnendruck 26–30 mmHg) 1)
Lin et al. (2021) berichteten über ein 5-jähriges Mädchen mit neuartigen compound-heterozygoten Mutationen im LTBP2-Gen (c.3672delC, c.3542delT). 1) Diese Mutationen waren neu und in der GnomAD East Asian Database nicht registriert. Die Anwendung in der Pränataldiagnostik und genetischen Beratung wird als zukünftige Richtung aufgezeigt.
Al Motawa et al. (2021) berichteten über eine Familie mit homozygoter ADAMTS10 c.2050C>T p(Arg684*)-Mutation und betonten die Bedeutung der genetischen Beratung und Präimplantationsdiagnostik. 2) Die Eltern der Patientin waren blutsverwandt (Cousinenehe).
Die Evidenz für eine Wachstumshormontherapie bei Kleinwuchs im Rahmen des Weil-Marchesani-Syndroms ist derzeit nicht etabliert. In dem von Al Motawa et al. berichteten Fall war der Wachstumshormon-Peak mit 7,89 ng/mL nicht niedrig, dennoch wurde eine probatorische Wachstumshormontherapie begonnen. 2) Zur Bestätigung von Wirksamkeit und Sicherheit sind weitere Untersuchungen erforderlich.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.