Weill-Marchesani Syndrome (WMS) is a hereditary connective tissue disorder. It is also called spherophakia-brachymorphia syndrome or mesodermal dysmorphodystrophy. Alternative names include Marchesani syndrome and inverted Marfan syndrome. It was first reported by Weill and Marchesani over 80 years ago. 2)
The prevalence is estimated at 1 in 100,000 people. 1) Inheritance can be either autosomal recessive (AR) or autosomal dominant (AD), with 45% autosomal recessive, 39% autosomal dominant, and 16% sporadic. 1)
It is classified into four subtypes based on the causative gene.
WMS1: ADAMTS10 (autosomal recessive) 2)
WMS2: FBN1 (autosomal dominant)
WMS3: LTBP2 (autosomal recessive) 1)
WMS4: ADAMTS17 (autosomal recessive)
It is characterized by a body type opposite to Marfan syndrome and is also called “Inverted Marfan syndrome.” While Marfan syndrome presents with tall stature, arachnodactyly, and upward lens dislocation, Weill-Marchesani syndrome presents with short stature, brachydactyly, and downward lens dislocation.
QHow rare is Weill-Marchesani syndrome?
A
It is a rare disease with an estimated prevalence of 1 in 100,000 people. 1) The inheritance pattern is autosomal recessive in 45%, autosomal dominant in 39%, and sporadic in 16%. Four causative genes are known: ADAMTS10, FBN1, LTBP2, and ADAMTS17.
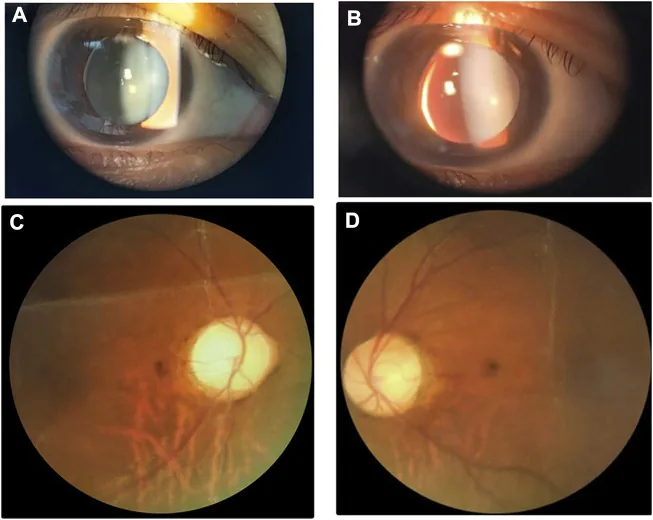
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
Slit-lamp and fundus images showing a small thick lens with inferonasal subluxation. The image is useful for recognizing the lens abnormalities that overlap with Weill-Marchesani syndrome.
Ocular and systemic findings are summarized below. The frequency of each finding is based on statistics from 128 cases by Faivre (2003). 2)
Ophthalmic Findings
Microspherophakia: The equatorial diameter is smaller than normal, and the lens appears spherical. One case measured LT (lens thickness) of 5.36 mm. Present in 84% of cases. 1)
Lens dislocation: Present in 73% of cases, often downward. May be accompanied by iridodonesis.
High myopia: Present in 94% of cases. Reports have reached OD -19.00 DS and OS -19.50 DS. 1)
Secondary glaucoma: Present in 80% of cases. Primarily angle-closure glaucoma due to pupillary block. 2)
Shallow anterior chamber: Presents with an anterior chamber depth of approximately 2 CT (corneal thickness). 1)
When the spherophakia moves forward, pupil block occurs, leading to angle-closure glaucoma. 2) It is reported that 80% of patients with Weill-Marchesani syndrome develop glaucoma. Miotics are contraindicated because they worsen this mechanism (see “Standard Treatment” section for details).
The cause of Weill-Marchesani syndrome is mutations in genes encoding extracellular matrix (ECM) components.
FBN1 (fibrillin-1): Autosomal dominant inheritance. Encodes a major component of microfibrils and is involved in the structure of the zonule of Zinn.
ADAMTS10 and ADAMTS17: Autosomal recessive inheritance. Known to be closely associated with fibrillin-1; their deficiency leads to Weill-Marchesani syndrome-like symptoms.
LTBP2 (Latent TGF-β Binding Protein 2): Autosomal recessive inheritance. Involved in extracellular matrix production and is present in elastic tissues and the ciliary zonule. 1)
Specific mutation examples include compound heterozygous mutations c.3672delC and c.3542delT in the LTBP2 gene (not registered in the GnomAD East Asian database). 1) Additionally, a homozygous mutation c.2050C>T p(Arg684*) in ADAMTS10 has been reported. 2)
Consanguineous marriage is a risk factor, and a report from Saudi Arabia found that 57% of patients had a history of consanguineous marriage. 2)
The diagnosis of Weill-Marchesani syndrome is made clinically based on a combination of ophthalmic and systemic findings. Genetic testing is useful for confirmation.
QWhat is the difference between Marfan syndrome and Weill-Marchesani syndrome?
A
Weill-Marchesani syndrome presents with short stature, brachydactyly, and inferior lens dislocation, whereas Marfan syndrome presents with tall stature, arachnodactyly, and superotemporal lens dislocation. The contrasting body habitus has led to the term “Inverted Marfan syndrome.” Homocystinuria also causes lens dislocation (often inferonasal), but intellectual disability and thrombosis help in differentiation. Weill-Marchesani syndrome is associated with spherophakia, making glaucoma more common compared to other syndromes with lens dislocation.
The basic treatment strategy is a stepwise intervention according to the stage of the disease. In mild cases, observation is recommended; if glaucoma or lens complications progress, surgery should be considered. Surgery is recommended before the lens falls into the vitreous cavity.
Lens extraction + anterior vitrectomy + scleral-fixated intraocular lens (IOL) insertion: Reports show postoperative intraocular pressure improved to 13 mmHg in the right eye and 12 mmHg in the left eye. 2)
Physical therapy: Management of joint stiffness and brachydactyly.
Cardiac follow-up: Screening and management of cardiovascular defects (24%).
Growth hormone (GH) therapy: Evidence is not established and is at an experimental stage. There is a report of a trial initiation in a case with a GH peak of 7.89 ng/mL. 2)
QWhy are miotics contraindicated?
A
Miotics (cholinergic agents) contract the ciliary muscle and relax the zonules. As a result, the microspherophakic lens moves further forward, promoting pupillary block and worsening acute angle-closure glaucoma. In Weill-Marchesani syndrome, the principle of treatment is to use cycloplegics (mydriatics) to retract the lens.
The pathology of Weill-Marchesani syndrome is understood primarily as weakening of the zonules and lens capsule due to abnormalities in extracellular matrix components.
FBN1 mutations disrupt microfibril structure, causing zonular weakness and increased lens mobility. ADAMTS10 and ADAMTS17 are closely related to fibrillin-1, and their deficiency also produces Weill-Marchesani syndrome-like symptoms.
Normally, around the 5th to 6th month of gestation, the lens temporarily becomes spherical. It then changes to an elliptical shape due to normal mesodermal development, but if mesodermal abnormalities occur, this spherical shape is maintained, resulting in microspherophakia. 1)
AR type (ADAMTS10/LTBP2)
Inheritance pattern: Autosomal recessive
Main genes: ADAMTS10, LTBP2, ADAMTS17
Severity of ocular findings: Tends to be high (LTBP2: LT 5.36 mm, IOP 26–30 mmHg) 1)
Systemic findings: Short stature and brachydactyly are prominent
Lin et al. (2021) reported a 5-year-old girl with novel compound heterozygous mutations in the LTBP2 gene (c.3672delC, c.3542delT). 1) These mutations were novel and not registered in the GnomAD East Asian database. Application to prenatal diagnosis and genetic counseling is indicated as a future direction.
Al Motawa et al. (2021) reported a family with a homozygous ADAMTS10 c.2050C>T p(Arg684*) mutation and emphasized the importance of genetic counseling and preimplantation genetic diagnosis. 2) The patient’s parents were consanguineous (first cousins).
Growth hormone therapy for short stature associated with Weill-Marchesani syndrome currently lacks established evidence. In the case reported by Al Motawa et al., the growth hormone peak was 7.89 ng/mL, which was not low, but growth hormone therapy was initiated on a trial basis. 2) Further studies are needed to establish its efficacy and safety.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.