Weill-Marchesani sendromu (WMS), kalıtsal bir bağ dokusu hastalığıdır. Ayrıca Sferofaki-Brakimorfi sendromu veya Mezodermal dismorfo-distrofi olarak da adlandırılır. Diğer adları Marchesani sendromu ve Ters Marfan sendromudur. 80 yıldan uzun süre önce Weill ve Marchesani tarafından tanımlanmıştır. 2)
Görülme sıklığının 100.000’de 1 vaka olduğu tahmin edilmektedir. 1) Kalıtım şekli hem otozomal resesif (OR) hem de otozomal dominant (OD) olabilir; %45 otozomal resesif, %39 otozomal dominant ve %16 sporadik olduğu bildirilmiştir. 1)
Neden olan gene göre dört alt tipe ayrılır.
WMS1: ADAMTS10 (otozomal resesif) 2)
WMS2: FBN1 (otozomal dominant)
WMS3: LTBP2 (otozomal resesif) 1)
WMS4: ADAMTS17 (otozomal resesif)
Marfan sendromunun tersine bir vücut yapısı sergilemesiyle karakterizedir ve “Ters Marfan sendromu” olarak da adlandırılır. Marfan sendromu uzun boy, örümcek parmaklar ve lensin yukarıya doğru yer değiştirmesi ile kendini gösterirken, Weill-Marchesani sendromu kısa boy, kısa parmaklar ve lensin aşağıya doğru yer değiştirmesi ile kendini gösterir.
QWeill-Marchesani sendromu ne kadar nadir bir hastalıktır?
A
Prevalansı 100.000’de 1 vaka olarak tahmin edilen nadir bir hastalıktır. 1) Kalıtım şekli %45 otozomal resesif, %39 otozomal dominant ve %16 sporadiktir. ADAMTS10, FBN1, LTBP2 ve ADAMTS17 olmak üzere dört farklı sorumlu gen bilinmektedir.
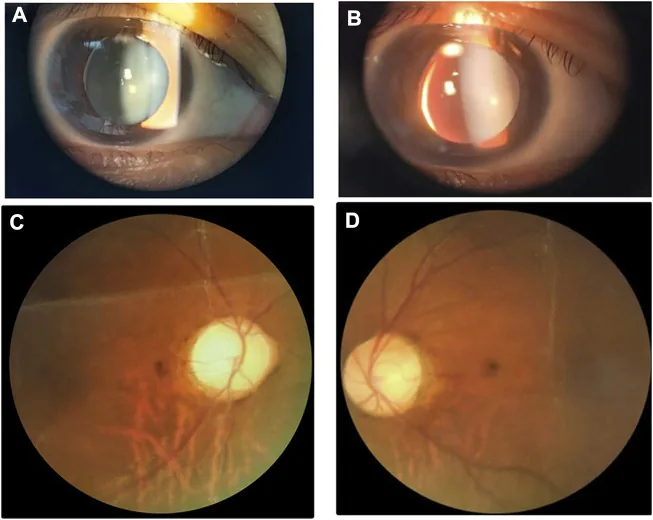
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
İnferonazal subluksasyonlu küçük kalın lensi gösteren yarık lamba ve fundus görüntüleri. Görüntü, Weill-Marchesani sendromu ile örtüşen lens anormalliklerini tanımak için faydalıdır.
Göz bulguları ve sistemik bulgular aşağıda özetlenmiştir. Her bir bulgunun sıklığı Faivre (2003)‘ün 128 vakalık istatistiğine dayanmaktadır. 2)
Oftalmolojik Bulgular
Mikrosferofaki: Ekvator çapı normalden daha küçük ve küresel şekildedir. LT (lens kalınlığı) 5.36 mm olarak ölçülmüş bir vaka vardır. %84’ünde görülür. 1)
Lens subluksasyonu: %73’ünde görülür ve sıklıkla aşağıya doğru yer değiştirir. İridodonez eşlik edebilir.
Yüksek miyopi: %94’ünde görülür. OD -19.00 DS ve OS -19.50 DS’ye ulaşan bildirilmiş vakalar vardır. 1)
Sekonder glokom: %80’inde görülür. Pupil bloğuna bağlı açı kapanması glokomu baskındır. 2)
Küresel lens öne doğru hareket ettiğinde pupil bloğu oluşur ve açı kapanması glokomu gelişir. 2) Weill-Marchesani sendromlu hastaların %80’inde glokom görüldüğü bildirilmiştir. Miyotik ilaçlar bu mekanizmayı kötüleştirdiği için kontrendikedir (ayrıntılar için “Standart Tedavi Yöntemleri” bölümüne bakın).
Weill-Marchesani sendromunun nedeni, hücre dışı matris (ECM) bileşenlerini kodlayan genlerdeki mutasyonlardır.
FBN1 (fibrillin-1): Otozomal dominant kalıtım. Mikrofibrillerin ana bileşenini kodlar ve Zinn zonüllerinin yapısında rol oynar.
ADAMTS10 ve ADAMTS17: Otozomal resesif kalıtım. Fibrillin-1 ile yakın ilişkili olduğu bilinir ve eksiklikleri Weill-Marchesani sendromu benzeri semptomlara yol açar.
LTBP2 (Latent TGF-β Bağlayıcı Protein 2): Otozomal resesif kalıtım. Hücre dışı matriks üretiminde rol alır ve elastik doku ile siliyer zonüllerde bulunur. 1)
Spesifik mutasyon örnekleri olarak, LTBP2 geninde c.3672delC ve c.3542delT bileşik heterozigot mutasyonları (GnomAD Doğu Asya veritabanında kayıtlı değil) rapor edilmiştir. 1) Ayrıca ADAMTS10 c.2050C>T p(Arg684*) homozigot mutasyonu da bildirilmiştir. 2)
Akraba evliliği risk faktörüdür ve Suudi Arabistan’dan bir raporda hastaların %57’sinde akraba evliliği öyküsü bulunmuştur. 2)
QMarfan sendromu ile Weil-Marchesani sendromu arasındaki fark nedir?
A
Weil-Marchesani sendromu kısa boy, kısa parmaklar ve lensin aşağıya doğru yer değiştirmesi ile karakterizeyken, Marfan sendromu uzun boy, örümcek benzeri parmaklar ve lensin yukarı ve dışa doğru yer değiştirmesi ile karakterizedir. Vücut yapılarındaki bu zıtlık nedeniyle “Ters Marfan sendromu” olarak da adlandırılır. Homosistinüri de lens yer değiştirmesine (genellikle içe ve aşağıya) neden olur, ancak zihinsel engellilik ve tromboz eşlik etmesi ayırıcı tanıda yardımcıdır. Weil-Marchesani sendromunda lens küresel olduğu için, diğer lens yer değiştirmesi sendromlarına kıyasla glokom daha sık görülür.
Tedavinin temel prensibi, hastalığın ilerleme derecesine göre aşamalı müdahaledir. Hafif vakalarda takip yeterliyken, glokom veya lens komplikasyonları ilerlediğinde cerrahi düşünülür. Lensin vitreus boşluğuna düşmesinden önce cerrahi yapılması önerilir.
Lens ekstraksiyonu + ön vitrektomi + skleral fiksasyonlu göz içi lens (GİL) yerleştirilmesi: Ameliyat sonrası göz içi basıncının sağda 13 mmHg, solda 12 mmHg’ye düştüğü bildirilmiştir. 2)
İntrakapsüler lens ekstraksiyonu: Mikrosferofaki için yapılır.
Fizik tedavi: Eklem sertliği ve brakidaktiliye yönelik müdahale.
Kardiyak takip: Kardiyovasküler defektlerin (%24) taranması ve yönetimi.
Büyüme hormonu (GH) tedavisi: Kanıt düzeyi kesin olmayıp deneysel aşamadadır. GH pik değeri 7.89 ng/mL olan bir vakada deneysel olarak başlandığı bildirilmiştir. 2)
QMiyotik ilaçlar neden kontrendikedir?
A
Miyotik ilaçlar (kolinerjik ajanlar) siliyer kası kasarak Zinn zonüllerini gevşetir. Bu, küresel lensin daha da öne hareket etmesine neden olarak pupil bloğunu artırır ve açı kapanması glokomu atağını kötüleştirme riski taşır. Weill-Marchesani sendromunda, sikloplejikler (midriyatikler) kullanılarak lensin geriye çekilmesi tedavinin temelidir.
Weill-Marchesani sendromunun patofizyolojisi, ekstraselüler matriks bileşenlerindeki anormalliklere bağlı olarak siliyer zonüller ve lens kapsülünün zayıflaması etrafında anlaşılır.
FBN1 mutasyonu, mikrofibrillerin yapısını bozarak Zinn zonüllerinin zayıflamasına ve lens hareketliliğinin artmasına neden olur. ADAMTS10 ve ADAMTS17, fibrillin-1 ile yakından ilişkilidir ve bu genlerin eksikliği de Weill-Marchesani sendromu benzeri semptomlara yol açar.
LTBP2, ekstraselüler matriksteki mikrofibril stabilitesinde rol oynar. Mutasyonu, Zinn zonülleri ve lens kapsülünün zayıflamasına, mikrosferofaki ve lens ektopisine neden olur. 1)
Normalde, embriyonik 5-6. aylarda lens geçici olarak küresel bir şekil alır. Daha sonra mezodermin normal gelişimi ile eliptik şekle dönüşür, ancak mezodermdeki bir anormallik nedeniyle bu küresel şekil korunursa mikrosferofaki oluşur. 1)
AR tipi (ADAMTS10/LTBP2)
Kalıtım şekli: Otozomal resesif
Başlıca genler: ADAMTS10, LTBP2, ADAMTS17
Göz bulgularının şiddeti: Yüksek eğilim (LTBP2: LT 5.36 mm, göz içi basıncı 26-30 mmHg) 1)
Sistemik bulgular: Kısa boy ve brakidaktili belirgindir
Lin ve ark. (2021), LTBP2 geninde yeni bir bileşik heterozigot mutasyon (c.3672delC ve c.3542delT) taşıyan 5 yaşında bir kız çocuğu bildirdi. 1) Bu mutasyonlar, GnomAD Doğu Asya veritabanında kayıtlı olmayan yeni mutasyonlardı. Doğum öncesi tanı ve genetik danışmanlıkta uygulanması gelecekteki yön olarak belirtilmiştir.
Al Motawa ve ark. (2021), ADAMTS10 c.2050C>T p(Arg684*) homozigot mutasyonu taşıyan bir aileyi rapor etmiş ve genetik danışmanlık ile preimplantasyon genetik tanının önemini vurgulamıştır.2) Hastanın ebeveynleri akraba evliliği (kuzen evliliği) yapmıştı.
Wail-Marquessani sendromuna bağlı boy kısalığında büyüme hormonu tedavisinin etkinliği henüz kanıtlanmamıştır. Al Motawa ve arkadaşlarının bildirdiği vakada büyüme hormonu pik değeri 7.89 ng/mL ile düşük olmamasına rağmen deneysel olarak büyüme hormonu tedavisi başlatılmıştır. 2) Etkinlik ve güvenliğin belirlenmesi için ileri çalışmalara ihtiyaç vardır.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.