Le syndrome de Weill-Marchesani (WMS) est une maladie héréditaire du tissu conjonctif. Il est également appelé syndrome de sphérophakie-brachymorphie ou dysmorphodystrophie mésodermique. D’autres noms incluent le syndrome de Marchesani et le syndrome de Marfan inversé. Il a été décrit il y a plus de 80 ans par Weill et Marchesani. 2)
La prévalence est estimée à 1 cas pour 100 000 personnes. 1) Les modes de transmission sont à la fois autosomique récessif (AR) et autosomique dominant (AD), avec 45 % de cas autosomiques récessifs, 39 % autosomiques dominants et 16 % sporadiques. 1)
Quatre sous-types sont classés en fonction du gène responsable.
WMS1 : ADAMTS10 (autosomique récessif) 2)
WMS2 : FBN1 (autosomique dominant)
WMS3 : LTBP2 (autosomique récessif) 1)
WMS4 : ADAMTS17 (autosomique récessif)
Il se caractérise par une morphologie opposée à celle du syndrome de Marfan, et est également appelé « syndrome de Marfan inversé ». Alors que le syndrome de Marfan se manifeste par une grande taille, une arachnodactylie et une luxation supérieure du cristallin, le syndrome de Weill-Marchesani se caractérise par une petite taille, une brachydactylie et une luxation inférieure du cristallin.
QQuelle est la rareté du syndrome de Weill-Marchesani ?
A
Il s’agit d’une maladie rare dont la prévalence est estimée à 1 cas pour 100 000 personnes. 1) Le mode de transmission est autosomique récessif dans 45 % des cas, autosomique dominant dans 39 % et sporadique dans 16 %. Quatre gènes causaux sont connus : ADAMTS10, FBN1, LTBP2 et ADAMTS17.
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
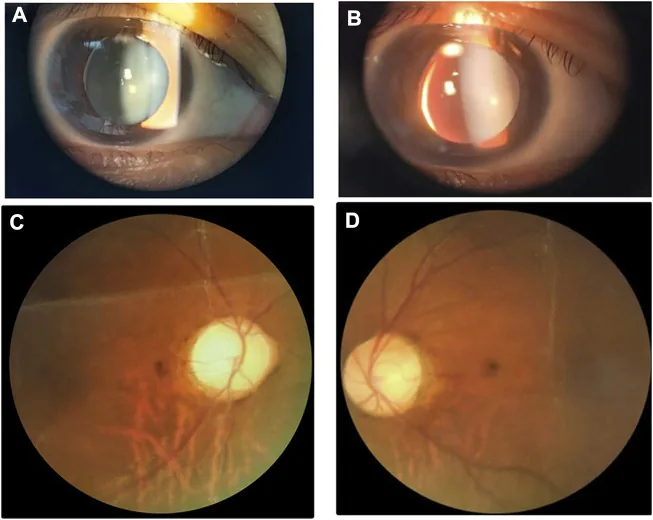
Images à la lampe à fente et du fond d’œil montrant un petit cristallin épais avec subluxation inféronasale. L’image est utile pour reconnaître les anomalies du cristallin qui se chevauchent avec le syndrome de Weill-Marchesani.
Les signes oculaires et généraux sont résumés ci-dessous. Les fréquences proviennent des statistiques de Faivre (2003) sur 128 cas. 2)
Signes ophtalmologiques
Microsphérophakie : cristallin de diamètre équatorial plus petit que la normale, de forme sphérique. Un cas mesurant 5,36 mm d’épaisseur (LT) a été rapporté. Présent dans 84 % des cas. 1)
Luxation du cristallin : observée dans 73 % des cas, souvent vers le bas. Peut s’accompagner d’iridodonésis.
Myopie forte : présente dans 94 % des cas. Des cas avec OD -19,00 DS et OS -19,50 DS ont été rapportés. 1)
Glaucome secondaire : présent dans 80 % des cas. Il s’agit principalement d’un glaucome par fermeture de l’angle dû à un bloc pupillaire. 2)
Lorsque le microsphérophakie se déplace vers l’avant, un bloc pupillaire se produit, entraînant un glaucome par fermeture de l’angle. 2) On estime que 80 % des patients atteints du syndrome de Weill-Marchesani développent un glaucome. Les myotiques sont contre-indiqués car ils aggravent ce mécanisme (voir la section « Traitement standard » pour plus de détails).
La cause du syndrome de Weill-Marchesani est une mutation des gènes codant pour les composants de la matrice extracellulaire (MEC).
FBN1 (fibrilline-1) : transmission autosomique dominante. Code pour le composant principal des microfibrilles, impliqué dans la structure de la zonule de Zinn.
ADAMTS10 et ADAMTS17 : transmission autosomique récessive. Connues pour leur association étroite avec la fibrilline-1, leur déficit entraîne des symptômes similaires au syndrome de Weill-Marchesani.
LTBP2 (Latent TGF-β Binding Protein 2) : transmission autosomique récessive. Impliquée dans la production de matrice extracellulaire, présente dans les tissus élastiques et la zonule ciliaire. 1)
Des exemples de mutations spécifiques incluent les mutations composées hétérozygotes c.3672delC et c.3542delT du gène LTBP2 (non enregistrées dans la base de données GnomAD Asie de l’Est) ont été rapportées. 1) De plus, une mutation homozygote c.2050C>T p(Arg684*) dans ADAMTS10 a également été rapportée. 2)
La consanguinité est un facteur de risque ; dans un rapport d’Arabie saoudite, 57 % des patients avaient des antécédents de consanguinité. 2)
Le diagnostic du syndrome de Weill-Marchesani est clinique, basé sur la combinaison des signes ophtalmologiques et systémiques. Le test génétique est utile pour la confirmation.
OCT du segment antérieur / microscopie ultrasonique (UBM) : utilisée pour évaluer la forme et la position du cristallin ainsi que l’angle iridocornéen.
IOLMaster / échographie en mode A : utile pour mesurer l’épaisseur du cristallin (ex. : LT 5,36 mm). 1)
Tonométrie : exemples de pression intraoculaire : OD 26,5 mmHg, OS 30,6 mmHg. 1)
QQuelle est la différence entre le syndrome de Marfan et le syndrome de Weill-Marchesani ?
A
Le syndrome de Weill-Marchesani se caractérise par une petite taille, une brachydactylie et un déplacement inférieur du cristallin, tandis que le syndrome de Marfan se manifeste par une grande taille, une arachnodactylie et un déplacement supéro-externe du cristallin. En raison de cette opposition morphologique, il est également appelé « syndrome de Marfan inversé ». L’homocystinurie peut aussi provoquer un déplacement du cristallin (souvent en bas et en dedans), mais la présence de déficience intellectuelle et de thrombose aide au diagnostic différentiel. De plus, le syndrome de Weill-Marchesani étant associé à une microsphérophakie, il entraîne plus fréquemment un glaucome que les autres syndromes avec ectopie du cristallin.
Le principe fondamental du traitement est une intervention progressive en fonction du stade d’avancement de la pathologie. En cas de forme légère, une simple surveillance est recommandée ; en cas de glaucome ou de complications cristalliniennes évoluées, une intervention chirurgicale est envisagée. Il est recommandé d’opérer avant que le cristallin ne tombe dans le vitré.
Extraction du cristallin + vitrectomie antérieure + implantation d’un cristallin artificiel fixé à la sclère (IOL) : des rapports indiquent une amélioration de la pression intraoculaire postopératoire à 13 mmHg à droite et 12 mmHg à gauche. 2)
Extraction intracapsulaire du cristallin : réalisée pour le microsphérophakie.
Trabéculectomie : réalisée en cas de mauvais contrôle du glaucome, avec des rapports indiquant qu’une intervention chirurgicale est nécessaire dans la moitié des cas. 2)
Correction de la réfraction et gestion de l’amblyopie
Kinésithérapie : prise en charge des raideurs articulaires et de la brachydactylie.
Suivi cardiaque : dépistage et prise en charge des anomalies cardiovasculaires (24 %).
Traitement par hormone de croissance (GH) : les preuves ne sont pas établies et le traitement est encore expérimental. Un cas avec un pic de GH à 7,89 ng/mL a été traité à titre expérimental. 2)
QPourquoi les myotiques sont-ils contre-indiqués ?
A
Les myotiques (cholinergiques) contractent le muscle ciliaire et relâchent la zonule de Zinn. En conséquence, le cristallin sphérique se déplace davantage vers l’avant, favorisant le bloc pupillaire et risquant d’aggraver une crise de glaucome par fermeture de l’angle. Dans le syndrome de Weill-Marchesani, le principe du traitement est d’utiliser des cycloplégiques (mydriatiques) pour faire reculer le cristallin.
La physiopathologie du syndrome de Weill-Marchesani repose sur la fragilisation de la zonule ciliaire et de la capsule du cristallin due à des anomalies des composants de la matrice extracellulaire.
Les mutations de FBN1 perturbent la structure des microfibrilles, entraînant une fragilisation de la zonule de Zinn et une hypermobilité du cristallin. ADAMTS10 et ADAMTS17 sont étroitement liés à la fibrilline-1, et leur déficit provoque également des symptômes similaires au syndrome de Weill-Marchesani.
LTBP2 est impliqué dans la stabilité des microfibrilles de la matrice extracellulaire. Les mutations entraînent une fragilisation de la zonule de Zinn et de la capsule du cristallin, provoquant un cristallin sphérique et une ectopie du cristallin. 1)
Mécanisme de développement du cristallin sphérique
Normalement, vers le 5e ou 6e mois de gestation, le cristallin devient temporairement sphérique. Ensuite, grâce au développement normal du mésoderme, il devient ellipsoïde. Cependant, une anomalie du mésoderme peut maintenir cette forme sphérique, conduisant à un microsphérophakie. 1)
Type AR (ADAMTS10/LTBP2)
Mode de transmission : autosomique récessif
Principaux gènes : ADAMTS10, LTBP2, ADAMTS17
Sévérité des signes oculaires : tendance élevée (LTBP2 : LT 5,36 mm, pression intraoculaire 26-30 mmHg) 1)
Signes systémiques : petite taille et brachydactylie marquées
Signes systémiques : Petite taille, brachydactylie, raideur articulaire
De plus, une hypothèse a été proposée selon laquelle une distribution anormale de l’actine serait impliquée dans la pathogénie du syndrome de Weill-Marchesani. 2)
7. Recherches récentes et perspectives futures (études en cours)
Lin et al. (2021) ont rapporté le cas d’une fillette de 5 ans présentant de nouvelles mutations hétérozygotes composites du gène LTBP2 (c.3672delC et c.3542delT). 1) Ces mutations étaient nouvelles et non répertoriées dans la base de données GnomAD Asie de l’Est. Leur application au diagnostic prénatal et au conseil génétique est envisagée comme une orientation future.
Al Motawa et al. (2021) ont rapporté une famille porteuse de la mutation homozygote ADAMTS10 c.2050C>T p(Arg684*), soulignant l’importance du conseil génétique et du diagnostic génétique préimplantatoire. 2) Les parents du patient étaient consanguins (mariage entre cousins).
L’hormonothérapie de croissance pour la petite taille associée au syndrome de Weill-Marchesani n’a pas encore de preuves établies à ce jour. Dans le cas rapporté par Al Motawa et al., le pic d’hormone de croissance était de 7,89 ng/mL, ce qui n’était pas faible, mais un traitement hormonal de croissance a été initié à titre expérimental. 2) Des études futures sont nécessaires pour établir son efficacité et sa sécurité.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.