Hội chứng Weill-Marchesani (WMS) là một bệnh di truyền của mô liên kết. Còn được gọi là hội chứng Spherophakia-Brachymorphia hoặc loạn sản trung bì (Mesodermal dysmorphodystrophy). Tên khác bao gồm hội chứng Marchesani, hội chứng Marfan đảo ngược. Được báo cáo bởi Weill và Marchesani hơn 80 năm trước. 2)
Tỷ lệ mắc bệnh ước tính khoảng 1 trên 100.000 người. 1) Kiểu di truyền có thể là lặn nhiễm sắc thể thường (AR) hoặc trội nhiễm sắc thể thường (AD), với tỷ lệ lần lượt là 45% lặn, 39% trội và 16% đơn lẻ. 1)
Dựa trên gen gây bệnh, hội chứng được phân thành 4 phân nhóm.
WMS1: ADAMTS10 (lặn nhiễm sắc thể thường) 2)
WMS2: FBN1 (trội nhiễm sắc thể thường)
WMS3: LTBP2 (lặn nhiễm sắc thể thường) 1)
WMS4: ADAMTS17 (lặn nhiễm sắc thể thường)
Đặc điểm là có thể hình trái ngược với hội chứng Marfan, còn được gọi là “Hội chứng Marfan đảo ngược”. Trong khi hội chứng Marfan biểu hiện chiều cao, ngón tay hình nhện và lệch thủy tinh thể lên trên, thì hội chứng Weill-Marchesani lại biểu hiện chiều thấp, ngón tay ngắn và lệch thủy tinh thể xuống dưới.
QHội chứng Weill-Marchesani hiếm gặp đến mức nào?
A
Đây là bệnh hiếm gặp với tỷ lệ mắc ước tính 1 ca trên 100.000 người. 1) Kiểu di truyền gồm di truyền lặn nhiễm sắc thể thường 45%, di truyền trội nhiễm sắc thể thường 39% và đơn lẻ 16%. Bốn gen gây bệnh đã được biết đến là ADAMTS10, FBN1, LTBP2 và ADAMTS17.
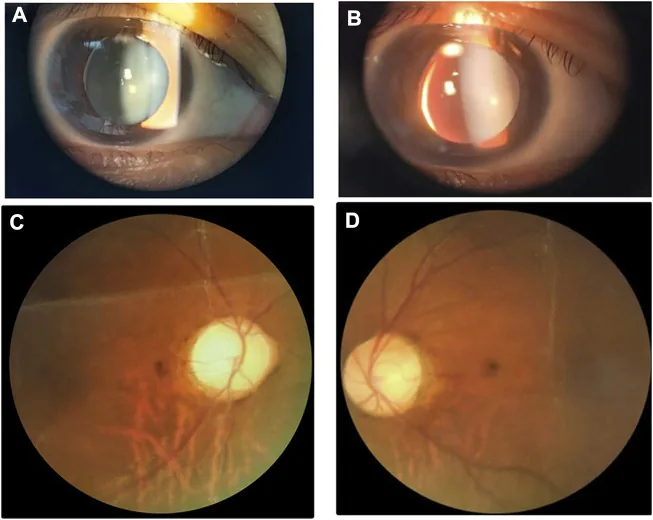
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
Hình ảnh đèn khe và đáy mắt cho thấy thủy tinh thể nhỏ dày với lệch dưới trong mũi. Hình ảnh hữu ích để nhận biết các bất thường thủy tinh thể chồng lấn với hội chứng Weill-Marchesani.
Các dấu hiệu ở mắt và toàn thân được trình bày dưới đây. Tần suất các dấu hiệu dựa trên thống kê 128 ca của Faivre (2003). 2)
Dấu hiệu nhãn khoa
Thể thủy tinh hình cầu nhỏ: Đường kính xích đạo nhỏ hơn bình thường và có dạng hình cầu. Có trường hợp đo được LT (độ dày thể thủy tinh) 5,36 mm. Gặp ở 84%. 1)
Lệch thể thủy tinh: Gặp ở 73%, thường lệch xuống dưới. Có thể kèm theo rung mống mắt.
Cận thị nặng: Gặp ở 94%. Có báo cáo trường hợp đạt OD -19,00 DS và OS -19,50 DS. 1)
Glaucoma thứ phát: gặp ở 80%. Chủ yếu là glaucoma góc đóng do block đồng tử. 2)
Hóa lỏng dịch kính và tăng độ dày giác mạc cũng được báo cáo.
Biểu hiện toàn thân
Chiều cao thấp: gặp ở 98%. Có báo cáo chiều cao 103 cm (Z-score -5.4). 2)
Ngón tay ngắn và cong: gặp ở 98%. 2)
Cứng khớp: gặp ở 62%. 2)
Dày da, giả vóc dáng cơ bắp: Có hình dáng cơ thể đặc trưng.
Bất thường khuôn mặt: Khe mi hẹp, lông mi dài, sống mũi rộng, môi trên mỏng. 2)
Khuyết tật tim mạch: Gặp ở 24%. Bao gồm còn ống động mạch, v.v. 2)
Khuyết tật trí tuệ: Gặp ở 13%. 2)
Khiếm thính cũng được báo cáo.
Dưới đây tóm tắt tần suất các triệu chứng chính.
Triệu chứng
Tần suất (%)
Chiều cao thấp
98
Ngón tay ngắn
98
Cận thị
94
Thể thủy tinh hình cầu nhỏ
84
Glôcôm thứ phát
80
Lệch thể thủy tinh
73
Cứng khớp
62
Khuyết tật tim mạch
24
Đục thủy tinh thể
23
Thiểu năng trí tuệ
13
QTại sao bệnh tăng nhãn áp xảy ra?
A
Khi thể thủy tinh hình cầu nhỏ di chuyển về phía trước, sẽ gây ra block đồng tử và dẫn đến tăng nhãn áp góc đóng. 2) Khoảng 80% bệnh nhân hội chứng Weill-Marchesani bị tăng nhãn áp. Thuốc co đồng tử làm trầm trọng thêm cơ chế này nên chống chỉ định (xem chi tiết tại mục “Phương pháp điều trị tiêu chuẩn”).
Nguyên nhân của hội chứng Weill-Marchesani là đột biến gen mã hóa các thành phần của chất nền ngoại bào (ECM).
FBN1 (Fibrillin-1): Di truyền trội nhiễm sắc thể thường. Mã hóa thành phần chính của sợi nhỏ, tham gia cấu trúc dây chằng Zinn.
ADAMTS10 và ADAMTS17: Di truyền lặn nhiễm sắc thể thường. Có liên quan chặt chẽ với fibrillin-1, thiếu hụt gây triệu chứng giống hội chứng Weill-Marchesani.
LTBP2 (Protein liên kết TGF-β tiềm ẩn 2): Di truyền lặn nhiễm sắc thể thường. Tham gia sản xuất chất nền ngoại bào, hiện diện ở mô đàn hồi và dây chằng thể mi. 1)
Ví dụ đột biến cụ thể: đột biến dị hợp tử kép c.3672delC và c.3542delT trên gen LTBP2 (chưa đăng ký trong cơ sở dữ liệu GnomAD Đông Á) đã được báo cáo. 1) Ngoài ra, đột biến đồng hợp tử ADAMTS10 c.2050C>T p(Arg684*) cũng đã được báo cáo. 2)
Hôn nhân cận huyết là yếu tố nguy cơ; một báo cáo từ Ả Rập Xê Út cho thấy 57% bệnh nhân có tiền sử hôn nhân cận huyết. 2)
Chẩn đoán hội chứng Weill-Marchesani được thực hiện lâm sàng dựa trên kết hợp các dấu hiệu nhãn khoa và toàn thân. Xét nghiệm di truyền hữu ích để xác định chẩn đoán.
QSự khác biệt giữa hội chứng Marfan và hội chứng Weill-Marchesani là gì?
A
Hội chứng Weill-Marchesani biểu hiện với vóc người thấp, ngón tay ngắn và lệch thể thủy tinh xuống dưới, trong khi hội chứng Marfan biểu hiện với vóc người cao, ngón tay dài như chân nhện và lệch thể thủy tinh lên trên và ra ngoài. Do sự tương phản về hình thể, hội chứng này còn được gọi là “Hội chứng Marfan đảo ngược”. Niệu niệu (homocystinuria) cũng gây lệch thể thủy tinh (thường lệch vào trong và xuống dưới), nhưng thiểu năng trí tuệ và huyết khối giúp phân biệt. Hội chứng Weill-Marchesani có thể thủy tinh hình cầu, do đó dễ bị glôcôm hơn các hội chứng lệch thể thủy tinh khác.
Nguyên tắc điều trị cơ bản là can thiệp theo từng giai đoạn dựa trên mức độ tiến triển của bệnh. Trường hợp nhẹ thì theo dõi, nếu glôcôm hoặc biến chứng thể thủy tinh tiến triển thì cân nhắc phẫu thuật. Nên phẫu thuật trước khi thể thủy tinh rơi vào dịch kính.
Phẫu thuật lấy thể thủy tinh + cắt dịch kính trước + đặt thủy tinh thể nhân tạocố định củng mạc (IOL): Có báo cáo cải thiện nhãn áp sau phẫu thuật lần lượt là 13 mmHg mắt phải và 12 mmHg mắt trái. 2)
Phẫu thuật lấy thể thủy tinh trong bao: Được thực hiện đối với thể thủy tinh hình cầu nhỏ.
Cắt mống mắt chu biên / Mở mống mắt chu biên bằng laser (LPI): Dùng để giải phóng block đồng tử. 2)
Phẫu thuật cắt bè củng mạc: Được thực hiện ở những trường hợp glôcôm kiểm soát kém, có báo cáo cho thấy một nửa số ca cần phẫu thuật. 2)
Theo dõi tim: Sàng lọc và quản lý các khuyết tật tim mạch (24%).
Liệu pháp hormone tăng trưởng (GH): Bằng chứng chưa được thiết lập, đang ở giai đoạn thử nghiệm. Có báo cáo về việc bắt đầu điều trị thử nghiệm ở một bệnh nhân có đỉnh GH 7,89 ng/mL. 2)
QTại sao thuốc co đồng tử lại chống chỉ định?
A
Thuốc co đồng tử (thuốc cholinergic) làm co cơ thể mi và giãn dây chằng Zinn. Kết quả là thể thủy tinh hình cầu nhỏ di chuyển về phía trước, làm tăng block đồng tử và có nguy cơ làm nặng thêm cơn glôcôm góc đóng. Trong hội chứng Weil-Marchesani, nguyên tắc điều trị là dùng thuốc liệt điều tiết (thuốc giãn đồng tử) để đẩy thể thủy tinh ra sau.
Bệnh sinh của hội chứng Weil-Marchesani được hiểu là do sự yếu kém của dây chằng Zinn và bao thể thủy tinh do bất thường các thành phần của chất nền ngoại bào.
Đột biến FBN1 làm tổn thương cấu trúc sợi nhỏ, gây yếu dây chằng Zinn và tăng khả năng di động của thể thủy tinh. ADAMTS10 và ADAMTS17 liên quan chặt chẽ với fibrillin-1; sự thiếu hụt các yếu tố này cũng gây ra các triệu chứng giống hội chứng Weil-Marchesani.
LTBP2 tham gia vào sự ổn định của sợi nhỏ trong chất nền ngoại bào. Đột biến làm yếu dây chằng Zinn và bao thể thủy tinh, dẫn đến thể thủy tinh hình cầu nhỏ và lệch thể thủy tinh. 1)
Bình thường, vào khoảng tháng thứ 5-6 của thai kỳ, thể thủy tinh tạm thời có hình cầu. Sau đó, nhờ sự phát triển bình thường của trung bì, nó chuyển thành hình bầu dục, nhưng nếu trung bì bất thường, hình cầu này được duy trì và dẫn đến thể thủy tinh hình cầu nhỏ. 1)
Thể AR (ADAMTS10/LTBP2)
Kiểu di truyền: Lặn nhiễm sắc thể thường
Gen chính: ADAMTS10, LTBP2, ADAMTS17
Mức độ nặng của triệu chứng mắt: Xu hướng cao (LTBP2: LT 5.36 mm, nhãn áp 26-30 mmHg) 1)
Lin và cộng sự (2021) đã báo cáo một bé gái 5 tuổi mang đột biến dị hợp tử kép mới trên gen LTBP2 (c.3672delC và c.3542delT). 1) Các đột biến này là đột biến mới chưa được đăng ký trong cơ sở dữ liệu GnomAD Đông Á. Ứng dụng trong chẩn đoán trước sinh và tư vấn di truyền được chỉ ra như một hướng đi trong tương lai.
Tư vấn di truyền trong gia đình có đột biến ADAMTS10
Al Motawa và cộng sự (2021) đã báo cáo một gia đình có đột biến đồng hợp tử ADAMTS10 c.2050C>T p(Arg684*) và nhấn mạnh tầm quan trọng của tư vấn di truyền và chẩn đoán di truyền tiền làm tổ. 2) Cha mẹ của bệnh nhân là kết hôn cận huyết (kết hôn anh em họ).
Liệu pháp hormone tăng trưởng cho tình trạng thấp lùn liên quan đến hội chứng Weil-Marchesani hiện chưa có bằng chứng xác lập. Trong trường hợp được báo cáo bởi Al Motawa và cộng sự, đỉnh hormone tăng trưởng là 7,89 ng/mL, không thấp, nhưng liệu pháp hormone tăng trưởng đã được bắt đầu thử nghiệm. 2) Cần có nghiên cứu thêm để xác định hiệu quả và độ an toàn.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.