La sindrome di Weill-Marchesani (WMS) è una malattia ereditaria del tessuto connettivo. È anche chiamata sindrome sferofachia-brachimorfia o dismorfo-distrofia mesodermica. Altri nomi includono sindrome di Marchesani e sindrome di Marfan invertita. Fu descritta oltre 80 anni fa da Weill e Marchesani. 2)
La prevalenza è stimata in 1 caso ogni 100.000 persone. 1) La modalità di trasmissione può essere sia autosomica recessiva (AR) che autosomica dominante (AD), con una distribuzione del 45% autosomica recessiva, 39% autosomica dominante e 16% sporadica. 1)
In base al gene causativo, si classificano quattro sottotipi.
WMS1: ADAMTS10 (autosomica recessiva) 2)
WMS2: FBN1 (autosomica dominante)
WMS3: LTBP2 (autosomica recessiva) 1)
WMS4: ADAMTS17 (autosomica recessiva)
È caratterizzato da una corporatura opposta a quella della sindrome di Marfan, ed è anche chiamato “sindrome di Marfan invertita”. Mentre la sindrome di Marfan presenta alta statura, aracnodattilia e lussazione superiore del cristallino, la sindrome di Weill-Marchesani presenta bassa statura, brachidattilia e lussazione inferiore del cristallino.
QQuanto è rara la sindrome di Weill-Marchesani?
A
È una malattia rara con una prevalenza stimata di 1 caso ogni 100.000 persone. 1) La modalità di trasmissione è autosomica recessiva nel 45%, autosomica dominante nel 39% e sporadica nel 16%. Sono noti quattro geni causali: ADAMTS10, FBN1, LTBP2 e ADAMTS17.
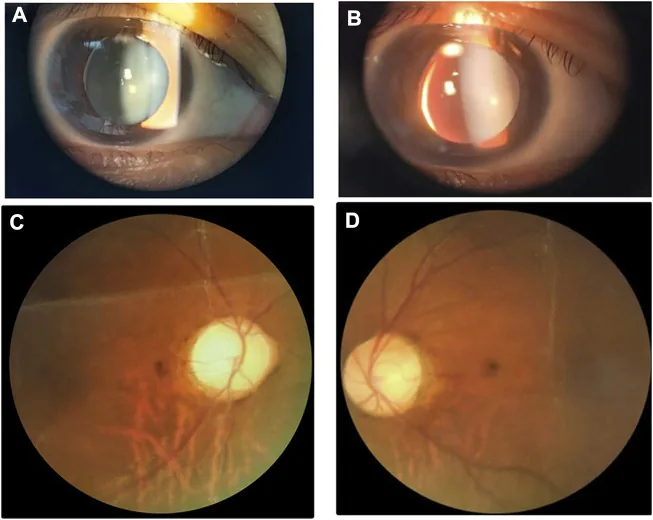
Li M, Li Y, Liu H, et al. Case report: A homozygous ADAMTSL2 missense variant causes geleophysic dysplasia with high similarity to Weill-Marchesani syndrome. Front Genet. 2022;13:1014188. Figure 1. PMCID: PMC9554500. License: CC BY.
Immagini con lampada a fessura e del fundus che mostrano un cristallino piccolo e spesso con sublussazione inferonasale. L’immagine è utile per riconoscere le anomalie del cristallino che si sovrappongono alla sindrome di Weill-Marchesani.
I reperti oculari e sistemici sono riassunti di seguito. Le frequenze si basano sulle statistiche di 128 casi di Faivre (2003). 2)
Reperti oftalmologici
Microsferofachia: il diametro equatoriale è più piccolo del normale e il cristallino assume forma sferica. Esempio di misurazione: LT (spessore del cristallino) 5,36 mm. Presente nell’84% dei casi. 1)
Lussazione del cristallino: presente nel 73% dei casi, spesso verso il basso. Può essere associata a iridodonesi.
Miopia elevata: presente nel 94% dei casi. Sono stati riportati casi con OD -19,00 DS e OS -19,50 DS. 1)
Glaucoma secondario: presente nell’80% dei casi. Predomina il glaucoma ad angolo chiuso da blocco pupillare. 2)
Quando il cristallino sferofaco si sposta in avanti, si verifica un blocco pupillare che porta al glaucoma ad angolo chiuso. 2) Si stima che l’80% dei pazienti con sindrome di Weill-Marchesani sviluppi glaucoma. I miotici sono controindicati perché peggiorano questo meccanismo (vedere la sezione “Trattamento standard” per i dettagli).
La sindrome di Weill-Marchesani è causata da mutazioni nei geni che codificano per i componenti della matrice extracellulare (ECM).
FBN1 (fibrillina-1): ereditarietà autosomica dominante. Codifica il componente principale delle microfibrille, coinvolto nella struttura della zonula di Zinn.
ADAMTS10 e ADAMTS17: ereditarietà autosomica recessiva. Strettamente correlati alla fibrillina-1; la loro carenza causa sintomi simili alla sindrome di Weill-Marchesani.
LTBP2 (proteina legante TGF-β latente 2): ereditarietà autosomica recessiva. Coinvolto nella produzione della matrice extracellulare, presente nel tessuto elastico e nella zonula ciliare. 1)
Esempi specifici di mutazioni includono le mutazioni eterozigoti composte c.3672delC e c.3542delT nel gene LTBP2 (non registrate nel database GnomAD dell’Asia orientale). 1) È stata inoltre segnalata una mutazione omozigote ADAMTS10 c.2050C>T p(Arg684*). 2)
La consanguineità è un fattore di rischio; in uno studio saudita, il 57% dei pazienti aveva genitori consanguinei. 2)
La diagnosi della sindrome di Weill-Marchesani viene effettuata clinicamente combinando i reperti oculari e sistemici. Il test genetico è utile per la conferma.
OCT del segmento anteriore e microscopia ultrasonica biomicroscopica (UBM): utilizzati per valutare forma, posizione del cristallino e angolo della camera anteriore.
IOLMaster ed ecografia A-scan: utili per misurare lo spessore del cristallino (es. LT 5,36 mm). 1)
Tonometria: esempi con OD 26,5 mmHg e OS 30,6 mmHg. 1)
Le varianti candidate vengono identificate mediante sequenziamento dell’esoma o sequenziamento di nuova generazione (NGS) e confermate con il metodo Sanger. 1)
QQual è la differenza tra la sindrome di Marfan e la sindrome di Weill-Marchesani?
A
La sindrome di Weill-Marchesani si caratterizza per bassa statura, brachidattilia e lussazione inferiore del cristallino, mentre la sindrome di Marfan presenta alta statura, aracnodattilia e lussazione supero-esterna del cristallino. A causa della morfologia corporea opposta, viene anche chiamata “sindrome di Marfan inversa”. Anche l’omocistinuria causa lussazione del cristallino (spesso infero-interna), ma il ritardo mentale e la trombosi aiutano nella diagnosi differenziale. La sindrome di Weill-Marchesani, essendo associata a microsferofachia, tende a complicarsi più frequentemente con glaucoma rispetto ad altre sindromi con lussazione del cristallino.
La strategia terapeutica di base prevede un intervento graduale in base alla progressione della patologia. Nei casi lievi si opta per l’osservazione; in caso di glaucoma o complicanze del cristallino in progressione si valuta l’intervento chirurgico. Si raccomanda di operare prima che il cristallino cada nel vitreo.
Estrazione del cristallino + vitrectomia anteriore + impianto di lente intraoculare (IOL) sclero-fissata: è stato riportato un miglioramento della pressione intraoculare post-operatoria a 13 mmHg nell’occhio destro e 12 mmHg nel sinistro. 2)
Estrazione intracapsulare del cristallino: eseguita per il cristallino microsferofachico.
Nei bambini, per prevenire l’ambliopia, si esegue precocemente la correzione refrattiva (occhiali) e si procede all’intervento chirurgico nel momento opportuno.
Fisioterapia: per rigidità articolare e brachidattilia.
Follow-up cardiaco: screening e gestione dei difetti cardiovascolari (24%).
Terapia con ormone della crescita (GH): le evidenze non sono consolidate ed è ancora in fase sperimentale. È stato riportato un caso con picco di GH di 7,89 ng/mL in cui è stata iniziata in via sperimentale. 2)
QPerché i miotici sono controindicati?
A
I miotici (farmaci colinergici) contraggono il muscolo ciliare e rilassano la zonula di Zinn. Di conseguenza, il cristallino sferofachico si sposta ulteriormente in avanti, favorendo il blocco pupillare e rischiando di peggiorare un attacco di glaucoma ad angolo chiuso. Nella sindrome di Weil-Marchesani, il principio del trattamento è l’uso di cicloplegici (midriatici) per far arretrare il cristallino.
La patologia della sindrome di Weil-Marchesani è compresa principalmente come un indebolimento della zonula di Zinn e della capsula del cristallino dovuto ad anomalie dei componenti della matrice extracellulare.
Le mutazioni di FBN1 danneggiano la struttura delle microfibrille, causando indebolimento della zonula di Zinn e ipermobilità del cristallino. ADAMTS10 e ADAMTS17 sono strettamente correlati alla fibrillina-1, e la loro carenza produce sintomi simili alla sindrome di Weil-Marchesani.
LTBP2 è coinvolto nella stabilità delle microfibrille della matrice extracellulare. Le mutazioni indeboliscono la zonula di Zinn e la capsula del cristallino, portando a sferofachia ed ectopia lentis. 1)
Normalmente, intorno al 5°-6° mese di gestazione, il cristallino diventa temporaneamente sferico. Successivamente, grazie al normale sviluppo del mesoderma, si trasforma in una forma ellittica, ma se il mesoderma è anomalo, questa forma sferica viene mantenuta, portando a un microsferofachia. 1)
Tipo AR (ADAMTS10/LTBP2)
Modalità di trasmissione: autosomica recessiva
Geni principali: ADAMTS10, LTBP2, ADAMTS17
Gravità dei reperti oculari: tendenzialmente elevata (LTBP2: LT 5.36 mm, pressione intraoculare 26-30 mmHg) 1)
Reperti sistemici: bassa statura e brachidattilia marcate
Inoltre, è stata proposta l’ipotesi che un’anomalia nella distribuzione dell’actina sia coinvolta nella patogenesi della sindrome di Weill-Marchesani. 2)
7. Ricerche recenti e prospettive future (studi in fase di ricerca)
Lin et al. (2021) hanno riportato il caso di una bambina di 5 anni con nuove mutazioni eterozigoti composte nel gene LTBP2 (c.3672delC e c.3542delT). 1) Queste mutazioni non erano state precedentemente registrate nel database GnomAD dell’Asia orientale. L’applicazione alla diagnosi prenatale e al counseling genetico è indicata come direzione futura.
Al Motawa et al. (2021) hanno riportato una famiglia con la mutazione omozigote ADAMTS10 c.2050C>T p(Arg684*), sottolineando l’importanza della consulenza genetica e della diagnosi genetica preimpianto. 2) I genitori del paziente erano consanguinei (cugini di primo grado).
La terapia con ormone della crescita per la bassa statura associata alla sindrome di Weil-Marchesani non ha ancora evidenze consolidate al momento. Nel caso riportato da Al Motawa et al., il picco dell’ormone della crescita era di 7,89 ng/mL, non considerato basso, ma la terapia è stata avviata in via sperimentale. 2) Sono necessari ulteriori studi per stabilirne l’efficacia e la sicurezza.
Lin Z, Zhu M, Deng H. A Pedigree Report of a Rare Case of Weill-Marchesani Syndrome with New Compound Heterozygous LTBP2 Mutations. Risk Manag Healthc Policy. 2021;14:1785-1789.
Al Motawa MNA, Alreshidi FS, Alluwaim FA, et al. Weill-Marchesani Syndrome, a Rare Presentation of Severe Short Stature with Review of the Literature. Am J Case Rep. 2021;22:e930824.