El penfigoide de las mucosas ocular (ocular mucous membrane pemphigoid, OCP) es una enfermedad ampollosa autoinmune crónica progresiva caracterizada por cicatrización del epitelio mucoso, incluida la conjuntiva, causada por autoanticuerpos contra componentes de adhesión de la membrana basal. El penfigoide de las mucosas (mucous membrane pemphigoid, MMP) es un grupo de enfermedades sistémicas que pueden afectar múltiples mucosas como la cavidad oral, superficie ocular, nasofaringe, laringe, esófago y genitales. Aquellos con afectación ocular se denominan penfigoide ocular o penfigoide cicatricial ocular. No son infrecuentes los casos sin lesiones cutáneas.
En oftalmología, el OCP se considera una enfermedad representativa de la epiteliopatía queratoconjuntival cicatricial con deficiencia de células madre del epitelio corneal, y sigue una progresión gradual desde conjuntivitis crónica a simbléfaron y finalmente queratinización corneal.
Esta enfermedad es rara. Un estudio de vigilancia en el Reino Unido reportó una incidencia anual de conjuntivitis cicatricial de aproximadamente 1,3 por millón de habitantes, y la prevalencia de OCP se estima en alrededor de 1 por cada 10.000 a 50.000 personas5). Afecta predominantemente a personas mayores de 60 a 80 años, con una proporción mujer:hombre de aproximadamente 2:15). El sitio mucoso más común es la cavidad oral (aproximadamente 85%), seguido de los ojos (aproximadamente 65%), y alrededor del 50% de los pacientes con OCP tienen afectación extraocular como lesiones orales o cutáneas2).
Se presenta de forma insidiosa como conjuntivitis crónica bilateral, y el período de enfermedad a menudo se extiende desde varios años hasta más de una década. En las etapas iniciales, solo hay síntomas inespecíficos de conjuntivitis, por lo que con frecuencia se trata como conjuntivitis alérgica, conjuntivitis crónica o síndrome de ojo seco durante mucho tiempo. Una anamnesis cuidadosa y la evaluación con lámpara de hendidura teniendo en cuenta esta enfermedad son extremadamente importantes para el diagnóstico temprano y la mejora del pronóstico visual.
Visión general del penfigoide de las membranas mucosas (MMP)
Órganos diana: Mucosas con epitelio escamoso estratificado en todo el cuerpo, incluyendo cavidad oral, superficie ocular, nasofaringe, laringe, esófago y genitales.
Lesiones cutáneas: A menudo ausentes. Las erosiones y cicatrices son más prominentes que las ampollas.
Edad de aparición: 60 a 80 años. Predominio femenino.
Complicaciones sistémicas: Disfagia por estenosis esofágica, y la estenosis laríngea puede afectar el pronóstico vital2).
Características del penfigoide cicatricial ocular (OCP)
Naturaleza de la afectación ocular: Conjuntivitis crónica bilateral con cicatrización conjuntival progresiva.
Patología final: Simbléfaron, agotamiento de las células madre del epitelio corneal y queratinización de la superficie ocular.
Afectación extraocular: Aproximadamente el 50% de los casos presentan afectación de otros sitios como la cavidad oral y la piel2).
Progresión insidiosa: Puede ser asintomático durante mucho tiempo, y algunos casos se descubren debido a la pérdida de visión2).
Q¿Son la misma enfermedad el penfigoide de las membranas mucosas (MMP) y el penfigoide cicatricial ocular (OCP)?
A
El penfigoide de las membranas mucosas (MMP) es un término general para las enfermedades ampollosas autoinmunes que afectan múltiples membranas mucosas, incluyendo la cavidad oral, los ojos, la nasofaringe, la laringe, el esófago y los genitales. Entre estos, los casos con afectación ocular se denominan penfigoide cicatricial ocular (OCP). El OCP puede afectar solo los ojos o acompañarse de lesiones extraoculares como afectación oral o cutánea; aproximadamente el 50% de los casos tienen afectación extraocular2). Dado que las lesiones cutáneas a menudo son discretas, los oftalmólogos son con frecuencia los primeros en llegar al diagnóstico.
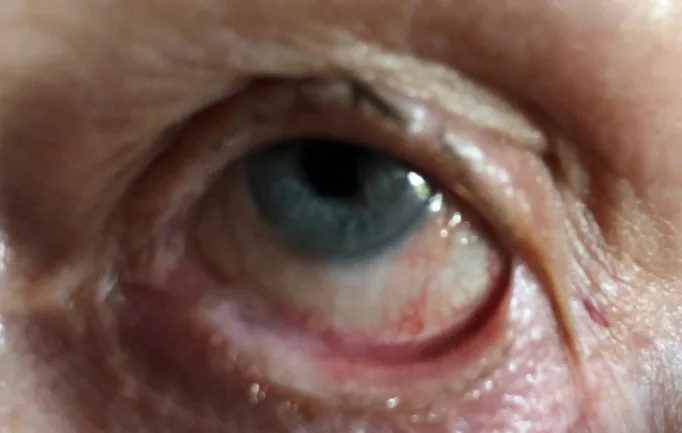
Stan C, Golea A, Gheorghe A, et al. Ocular cicatricial pemphigoid. Rom J Ophthalmol. 2020 Apr-Jun;64(2):226-230. Figure 2. PMCID: PMC7339695. License: CC BY.
En el ojo izquierdo, se forma simbléfaron (adhesión entre el párpado y la conjuntiva bulbar). Esto muestra un ejemplo específico de simbléfaron descrito en la sección de síntomas principales y hallazgos clínicos.
En la etapa inicial, los síntomas principales son hiperemia conjuntival crónica, sensación de sequedad, sensación de cuerpo extraño, ardor y lagrimeo. Estos son resistentes al tratamiento habitual del ojo seco, y su persistencia es un motivo para sospechar esta enfermedad 1). En casos avanzados, se añaden trastornos del movimiento ocular por simbléfaron, cierre palpebral incompleto, síntomas de irritación corneal por triquiasis, y disminución de la agudeza visual por opacidad corneal o invasión epitelial conjuntival.
La progresión de esta enfermedad es insidiosa, y puede permanecer asintomática durante mucho tiempo, con algunos casos descubiertos por primera vez cuando disminuye la agudeza visual2).
La clasificación de Foster (clasificación modificada de Tauber-Foster) se utiliza ampliamente para la estadificación. Progresa gradualmente desde la etapa I (conjuntivitis crónica), etapa II (acortamiento del fondo de saco inferior), etapa III (simbléfaron y vascularización corneal), hasta la etapa IV (queratinización de la superficie ocular) 2).
Etapa
Hallazgos principales
Etapa I
Conjuntivitis crónica, Rosa de Bengala positivo (deficiencia de mucina), fibrosis subepitelial conjuntival
Etapa II
Acortamiento del fondo de saco inferior (A–D: progresión evaluada en incrementos del 25%)
Etapa III
Simbléfaron, vascularización corneal, triquiasis, disminución de la secreción lagrimal
Etapa IV
Queratinización de la superficie ocular, anquilobléfaron, sequedad severa, dermatización corneal
En la etapa temprana, se observan cicatrices como tejido tendinoso blanco lineal o en parches en la conjuntiva palpebral. A medida que la enfermedad progresa, aparecen acortamiento del fondo de saco conjuntival, pérdida de las empalizadas de Vogt (POV), invasión epitelial conjuntival en la córnea, obstrucción del conducto lagrimal, pérdida de células caliciformes conjuntivales y ojo seco severo debido a la pérdida de las glándulas de Meibomio. Durante las exacerbaciones agudas, pueden ocurrir defectos epiteliales persistentes extensos.
Histológicamente, se observan fibrosis del estroma conjuntival, pérdida completa de células caliciformes y metaplasia escamosa 1). La pérdida de células caliciformes significa la pérdida de la función de secreción de mucina conjuntival, y la estabilidad de la película lagrimal se ve significativamente afectada. Además, la capa lipídica también se pierde debido a la destrucción de las glándulas de Meibomio, lo que aumenta la evaporación lagrimal y completa un ojo seco evaporativo severo.
Como complicaciones mucosas sistémicas, es necesario evaluar lesiones orales que causan gingivitis descamativa y lesiones ampollosas, lesiones esofágicas que causan disfagia, y lesiones laríngeas/traqueales que causan ronquera y disnea. La estenosis laríngea es un factor determinante del pronóstico vital que requiere manejo de la vía aérea, y se necesita colaboración urgente con un otorrinolaringólogo para pacientes con síntomas respiratorios 2). La estenosis esofágica causa disfagia y desnutrición, y se considera una evaluación endoscópica por un gastroenterólogo y dilatación con balón si es necesario. Las lesiones cutáneas a menudo aparecen como cambios eritematosos o atróficos en lugar de ampollas, y tienden a ocurrir particularmente en la cabeza, cara y cuello.
El penfigoide de las membranas mucosas oculares es causado por un mecanismo autoinmune (ver Sección 6 para detalles). Los factores de riesgo para el inicio incluyen envejecimiento, sexo femenino y antecedentes de otras enfermedades autoinmunes. No se ha identificado un desencadenante claro, pero se discute que la infección y la exposición a fármacos pueden estar involucradas en la ruptura de la tolerancia inmune. Se ha reportado una asociación con HLA-DQB1*0301 en estudios en el extranjero, lo que sugiere una predisposición genética.
Clínicamente, es importante diferenciar de otras enfermedades que presentan conjuntivitis cicatrizante. Las siguientes enfermedades tienen diferentes patologías y tratamientos, por lo que se debe reducir la causa en la primera visita.
Enfermedades autoinmunes/inflamatorias a diferenciar
Síndrome de Stevens-Johnson/necrólisis epidérmica tóxica: Trastorno mucocutáneo agudo debido a reacción farmacológica. Se diferencia por antecedentes de fiebre y erupción generalizada.
Enfermedad de injerto contra huésped (EICH): Ocurre después del trasplante de células madre hematopoyéticas. En casos de adherencia grave y progresiva, considere la comorbilidad con OCP4).
Sarcoidosis: Conjuntivitis granulomatosa no caseificante que puede cicatrizar. Reportada como una enfermedad imitadora de OCP3).
Liquen plano/dermatosis ampollosa IgA lineal: Requiere evaluación dermatológica.
Enfermedades exógenas a diferenciar
Seudopenfigoide inducido por fármacos: El uso prolongado de fármacos antiglaucomatosos (p. ej., latanoprost, timolol, brinzolamida) es un ejemplo típico. Mejora con la suspensión del fármaco 7).
Lesión química/Quemadura térmica: Las lesiones por álcali son particularmente graves.
Infecciosa: El tracoma es una causa principal en países en desarrollo. En países desarrollados, casos graves de queratoconjuntivitis adenoviral.
Iatrogénico: Antecedentes de cirugía oftálmica con incisión conjuntival
La inflamación crónica bilateral del epitelio corneal y conjuntival que progresa lentamente a cicatrización sugiere fuertemente OCP. Especialmente en mujeres mayores con triquiasis o entropión, evalúe cuidadosamente el acortamiento del fondo de saco conjuntival y la simbléfaron.
En la anamnesis, obtenga una historia detallada de traumatismos oculares, infecciones oculares y uso pasado y actual de gotas para los ojos y medicamentos orales. Además, realice una revisión sistémica de las lesiones mucosas en la cavidad oral, nasofaringe, laringe, esófago y piel, y colabore con dermatología, otorrinolaringología, gastroenterología y reumatología según sea necesario 2).
Citología por impresión: La citología de la conjuntiva confirma la pérdida de células caliciformes.
Tinción con fluoresceína: Se observa un aumento de la permeabilidad donde el epitelio conjuntival invade la superficie corneal, y junto con la pérdida de POV, indica deficiencia de células madre del epitelio corneal8).
El pilar del diagnóstico definitivo es la biopsia conjuntival. Se toma una muestra de la conjuntiva bulbar inferior y se realizan simultáneamente el examen histopatológico del tejido fijado en formalina y la inmunofluorescencia directa (DIF) del tejido fijado en solución de Michel 5).
Un hallazgo positivo de DIF en OCP es la deposición lineal de IgG, IgA, IgM y complemento C3 a lo largo de la zona de la membrana basal del epitelio conjuntival 5). Sin embargo, los falsos negativos no son infrecuentes. Las causas incluyen enfermedad en fase de reposo, pérdida completa de la membrana basal en enfermedad terminal, sitio de biopsia inadecuado y variación local de la respuesta inmune. En casos con fuerte sospecha clínica, un DIF negativo no descarta la enfermedad, y múltiples biopsias, incluida la rebiopsia del ojo contralateral, pueden mejorar la sensibilidad 4,5).
Autoanticuerpos: Los anticuerpos anti-BP180 (anticuerpos anti-colágeno tipo XVII) y los anticuerpos anti-laminina 332 (anticuerpos anti-laminina 5) se detectan en muchos pacientes con OCP. Estos autoanticuerpos desempeñan un papel central en la patogenia.
Pruebas para diagnóstico diferencial: VSG, hemograma completo, perfil metabólico completo, ANA, SS-A/SS-B (síndrome de Sjögren), ANCA (granulomatosis con poliangeítis), ECA/lisozima (sarcoidosis)
Uso prolongado de fármacos epiteliotóxicos como colirios antiglaucoma. Mejora tras la suspensión del fármaco causal 7)
EICH crónica
Antecedentes de trasplante de células madre hematopoyéticas. Sin embargo, en casos con adherencias graves, se debe prestar atención a la posible coexistencia con OCP4)
Incluso si la ECA/lisozima son normales, la sarcoidosis permanece en el diagnóstico diferencial; la inmunofluorescencia negativa es útil para excluir OCP3)
Q¿Por qué no se puede descartar la OCP incluso si la inmunofluorescencia directa de la biopsia conjuntival es negativa?
A
La prueba DIF no es completamente sensible y existen múltiples factores que pueden causar falsos negativos. Las principales causas incluyen la fase de reposo de la enfermedad, casos en los que la membrana basal ha desaparecido por completo en la etapa terminal, cuando el sitio de la biopsia es de un área con baja actividad y la variación local en la deposición inmune. También se han reportado casos con resultados diferentes entre el ojo derecho e izquierdo 5). Cuando los hallazgos clínicos sugieren fuertemente OCP, la sensibilidad puede mejorar mediante pruebas repetidas, incluida una nueva biopsia del ojo contralateral u otro sitio 4). La detección de anticuerpos séricos anti-BP180 y anti-laminina 332 también es útil como ayuda diagnóstica.
Los objetivos del tratamiento son detener la cicatrización progresiva, prevenir y corregir las complicaciones corneales y palpebrales, y aliviar los síntomas. Esta enfermedad es un trastorno autoinmune sistémico y la terapia local por sí sola no puede controlar su progresión. El pilar del tratamiento es la terapia inmunosupresora sistémica, y se recomienda encarecidamente la colaboración con los departamentos de dermatología y reumatología 2).
Los medicamentos se seleccionan de forma escalonada según la actividad de la enfermedad y el estado inflamatorio. En todos los casos, la monitorización de efectos secundarios y los análisis de sangre periódicos son esenciales 2,5).
Terapia sistémica: selección escalonada
Leve a moderado: Dapsona (diaminodifenilsulfona) 50–200 mg/día es de primera línea. Es obligatorio excluir la deficiencia de G6PD y monitorizar la anemia y la metahemoglobinemia 5).
Dapsona ineficaz/moderado: Escalar a micofenolato de mofetilo 1–3 g/día, azatioprina 1–2 mg/kg/día o metotrexato 7.5–25 mg/semana 2).
Grave/exacerbación aguda: Combinación de ciclofosfamida 1–2 mg/kg/día y prednisolona en dosis altas a corto plazo 5).
Tratamiento sintomático: Gotas oftálmicas de esteroides a baja concentración para antiinflamación, lágrimas artificiales sin conservantes para el manejo del ojo seco y depilación para reducir la irritación corneal.
Reconstrucción de simbléfaron: Después de controlar la inflamación, realizar lisis de adherencias y reconstrucción con trasplante de membrana amniótica o injerto de mucosa oral 2).
Casos con glaucoma: Cambiar a gotas oftálmicas sin conservantes como medida básica. También hay informes de casos de cirugía de glaucoma mínimamente invasiva (p. ej., stent de gel XEN) 6).
Etapa terminal: Trasplante de epitelio mucoso cultivado para reconstrucción del epitelio corneal, prótesis corneal (Boston KPro tipo 2, OOKP) en casos graves
Muchos de los fármacos no están cubiertos por el seguro para esta enfermedad, por lo que se requiere una explicación suficiente y consideración ética antes de su introducción. Tenga en cuenta que la ciclosporina oral y la ciclofosfamida tampoco están cubiertas por el seguro para esta enfermedad. La dapsona puede inducir hemólisis de glóbulos rojos y metahemoglobinemia, por lo que se debe medir la actividad de la glucosa-6-fosfato deshidrogenasa (G6PD) antes de la administración para descartar deficiencia. Después de iniciar el tratamiento, se deben evaluar periódicamente el hemograma, la función hepática y la función renal, y aumentar gradualmente la dosis. El micofenolato de mofetilo es relativamente bien tolerado y adecuado para uso a largo plazo, pero se debe realizar un cribado de infecciones considerando el riesgo de infecciones oportunistas. La ciclofosfamida es un potente inmunosupresor para casos graves, pero conlleva riesgos de mielosupresión, cistitis hemorrágica y carcinogenicidad a largo plazo, por lo que se debe controlar la dosis total. El rituximab, como anticuerpo monoclonal anti-CD20, se posiciona como terapia de rescate para OCP refractario, y es importante evaluar el riesgo de reactivación del virus de la hepatitis B antes de la administración2).
El tratamiento local es importante como terapia sintomática adjunta a la terapia sistémica. Las gotas oftálmicas de corticosteroides en baja concentración reducen la inflamación de la superficie ocular, y las lágrimas artificiales sin conservantes manejan el ojo seco. Las intervenciones quirúrgicas como la oclusión del punto lagrimal deben considerarse con precaución, ya que pueden acelerar la cicatrización.
El tratamiento quirúrgico solo debe realizarse cuando la inflamación esté completamente controlada. La cirugía bajo inflamación activa puede provocar una progresión rápida de cicatrización, adherencia y queratinización2).
Cirugía de cataratas: Si es necesario, realizar después de una antiinflamación suficiente con terapia inmunosupresora sistémica iniciada el día de la cirugía. Si se realiza sin un control antiinflamatorio adecuado, pueden progresar rápidamente las adherencias y la queratinización postoperatoriamente.
Reconstrucción del epitelio corneal: Para defectos epiteliales persistentes causados por exacerbación aguda, primero realizar tratamiento conservador con corticosteroides orales. Para casos refractarios, las opciones incluyen trasplante de lámina de epitelio corneal cultivado autólogo o alogénico, trasplante de epitelio de mucosa oral autólogo, etc. El trasplante de epitelio de mucosa oral autólogo está aprobado como atención médica avanzada en Japón.
Cirugía para recuperación visual en etapa terminal: En casos graves con invasión conjuntival y queratinización de la córnea, el tratamiento quirúrgico para la recuperación visual tiene un pronóstico pobre y generalmente no se realiza.
En enfermedades bilaterales progresivas como el OCP grave, las indicaciones del trasplante autólogo de células madre limbares (CLAu, CLET, SLET) son limitadas. Muchos casos son bilaterales y carecen de un ojo donante sano, por lo que se considera el trasplante alogénico o el trasplante de epitelio mucoso cultivado 9). Una revisión sistemática del trasplante autólogo de células madre limbares, principalmente para traumatismos químicos unilaterales, informó una tasa de éxito anatómico del 69% y funcional del 60% en 1.023 ojos, pero es poco probable que sea la primera opción en enfermedades autoinmunes bilaterales como el OCP9).
Q¿Cómo se puede realizar de forma segura la cirugía de cataratas en pacientes con OCP?
A
La cirugía en presencia de inflamación activa es un factor de riesgo importante para la progresión rápida de simbléfaron y queratinización corneal. Al planificar la cirugía, se debe usar terapia inmunosupresora sistémica (p. ej., esteroides, ciclosporina, ciclofosfamida) desde el día de la cirugía para suprimir completamente la inflamación, minimizar el daño físico intraoperatorio a la superficie ocular y continuar la inmunosupresión después de la cirugía. Si la cirugía se realiza con un control inflamatorio preoperatorio insuficiente, existe un alto riesgo de progresión rápida de adherencias y queratinización posoperatoria. La selección de la técnica quirúrgica, el manejo sistémico y la colaboración con dermatología y reumatología son extremadamente importantes 2).
El OCP es una enfermedad autoinmune tipo II (citotóxica) caracterizada por la producción de autoanticuerpos contra componentes de la membrana basal asociados a hemidesmosomas 5).
BP180 (colágeno tipo XVII): Una proteína transmembrana de los hemidesmosomas que conecta las células epidérmicas/epiteliales con la membrana basal. Los anticuerpos anti-BP180 son uno de los autoanticuerpos más frecuentemente detectados en pacientes con OCP.
Laminina 332 (laminina 5): Una proteína de adhesión en la membrana basal que une los hemidesmosomas con el colágeno tipo IV. Los anticuerpos anti-laminina 332 también se detectan en muchos pacientes con OCP.
También se han reportado otros anticuerpos contra la integrina α6β4 y otros componentes de la membrana basal.
Cuando los autoanticuerpos se unen a la membrana basal del epitelio conjuntival, se activa el sistema del complemento y se reclutan células inflamatorias. En la fase aguda, los eosinófilos y neutrófilos son los principales protagonistas de la inflamación, mientras que en la fase crónica predomina la infiltración linfocítica 5). Las citocinas y proteasas liberadas activan los fibroblastos subepiteliales conjuntivales, promoviendo la producción excesiva de matriz extracelular como colágeno, lo que conduce a fibrosis y cicatrización conjuntival. Las citocinas de tipo Th2 como el factor de crecimiento transformante β (TGF-β), la interleucina-4 (IL-4), IL-5 e IL-13 están sobreexpresadas en la lesión y se consideran los principales impulsores de la fibrosis. Un desequilibrio entre la metaloproteinasa de matriz-9 (MMP-9) y los inhibidores tisulares (TIMP) conduce a una remodelación descontrolada del tejido conjuntival, lo que también es característico de la patología.
Agotamiento de las células madre del epitelio corneal y queratinización corneal
La progresión gradual de la cicatrización conjuntival corresponde a la clasificación de Foster. La inflamación crónica persistente conduce a fibrosis subconjuntival (estadio I), acortamiento del fondo de saco conjuntival (estadio II), simbléfaron (estadio III) y, finalmente, pérdida de las palizadas de Vogt (POV) en el limbo corneal8). Dado que las células madre del epitelio corneal residen en las POV, su pérdida implica una falla en la regeneración normal del epitelio corneal. El agotamiento de las células madre del epitelio corneal conduce a la conjuntivalización, donde el epitelio conjuntival invade la superficie corneal, resultando en opacidad corneal, vascularización y, finalmente, queratinización similar a la piel (estadio IV) 8).
La enfermedad de injerto contra huésped ocular crónica (oGVHD) después del trasplante de células madre hematopoyéticas generalmente se presenta con inflamación crónica de la superficie ocular, ojo seco y fibrosis conjuntival leve. Sin embargo, cuando ocurren cicatrización grave y rápidamente progresiva y simbléfaron, se debe considerar la posibilidad de OCP4). Aunque oGVHD y OCP tienen características clínicas similares, sus estrategias de tratamiento difieren, por lo que la diferenciación mediante biopsia conjuntival e IFD es importante 4).
Q¿Qué es el agotamiento de las células madre del epitelio corneal en la OCP?
A
El limbo corneal contiene células madre responsables de la regeneración del epitelio corneal, observables como las palizadas de Vogt (POV). En la OCP, la inflamación crónica y la cicatrización se extienden al limbo, provocando la pérdida de las POV y las células madre. Cuando las células madre se agotan, el epitelio conjuntival invade la superficie corneal (conjuntivalización), haciendo imposible la regeneración normal del epitelio corneal. Esto resulta en opacidad corneal, vascularización y queratinización, lo que lleva a una discapacidad visual grave 8). Esta condición representa la vía final común de enfermedades graves de la superficie ocular observadas en el penfigoide de las mucosas ocular, el síndrome de Stevens-Johnson y las lesiones químicas.
7. Investigación más reciente y perspectivas futuras
Estudios de series de casos han demostrado la eficacia de rituximab e IVIG para el OCP refractario, y se están estableciendo como opciones de tratamiento para casos resistentes a la terapia inmunosupresora convencional 2). La administración de rituximab elimina las células B productoras de autoanticuerpos, y se han reportado casos que logran remisión clínica. La IVIG tiene efectos antiinflamatorios e inmunomoduladores y se posiciona como terapia combinada para casos graves o resistentes a otros fármacos. También se discute el potencial de nuevos inmunomoduladores como los inhibidores de JAK, y su administración oral, que facilita el manejo ambulatorio, genera expectativas para su futura aplicación clínica.
En el campo de la reconstrucción del epitelio corneal, el trasplante de láminas de epitelio corneal cultivado y el trasplante de láminas de epitelio mucoso oral autólogo se han aplicado clínicamente, y su desarrollo como medicina avanzada está progresando. Las láminas de epitelio corneal autólogo se preparan utilizando células epiteliales corneales del ojo sano, pero no se pueden usar en OCP con afectación bilateral. Por lo tanto, se seleccionan láminas de epitelio corneal alogénico utilizando córneas proporcionadas por bancos de ojos, o láminas de epitelio mucoso oral autólogo preparadas a partir de células de la mucosa oral del propio paciente. La mucosa oral autóloga no tiene riesgo de rechazo y se espera que proporcione estabilización de la superficie ocular a largo plazo. Se han desarrollado materiales portadores como membrana amniótica, pegamento de fibrina, placas de cultivo termosensibles y membranas de fluoruro de polivinilideno (PVDF), ampliando las opciones de técnicas quirúrgicas 8).
Para casos con glaucoma concurrente, la cirugía filtrante convencional es difícil debido a la cicatrización conjuntival, pero se han reportado casos exitosos de cirugía de glaucoma mínimamente invasiva (MIGS) con el stent de gel XEN. Después de controlar la inflamación de la superficie ocular, el stent se inserta mediante un abordaje ab interno, y se han demostrado casos que logran control de la presión intraocular sin gotas oftálmicas y remisión de la inflamación al año de la cirugía, lo que atrae la atención como un nuevo enfoque para el glaucoma complicado con enfermedad grave de la superficie ocular 6). Se espera que en el futuro se acumulen resultados a largo plazo mediante estudios multicéntricos colaborativos y se evalúe la respuesta al tratamiento utilizando biomarcadores.
Farrag A, Chan A, Tong L. Cicatricial Conjunctivitis and Concurrent Clinical Features: A Case Study. Clinical medicine insights. Case reports. 2022;15:11795476221100605. doi:10.1177/11795476221100605. PMID:35601266; PMCID:PMC9121463.
Razzak A, Ait Ammar H, Bouazza M, Elbelhadji M. Accidental Discovery of Ocular Cicatricial Pemphigoid. Cureus. 2025;17(1):e77425. doi:10.7759/cureus.77425. PMID:39949452; PMCID:PMC11823481.
Murati Calderon RA, López-Fontanet JJ, Ramirez Marquez E, Tavares Reigada AS, Oliver A. Sarcoidosis: A Mimicker of Ocular Cicatricial Pemphigoid. Cureus. 2025;17(10):e93862. doi:10.7759/cureus.93862. PMID:41194826; PMCID:PMC12584608.
Taketani Y, Dehghani S, Sinha S, Freitag SK, Papaliodis G, Foster S, et al. Concurrence of Ocular Cicatricial Pemphigoid in Chronic Ocular Graft-Versus-Host Disease. Cornea. 2024;43(3):387-390. doi:10.1097/ICO.0000000000003386. PMID:38128104; PMCID:PMC12703942.
Tesorero JCC, Sosuan GMN, Lim Bon Siong R. Ocular Cicatricial Pemphigoid in a Healthy Elderly Male Filipino Patient. Acta Med Philipp. 2025;59(18):117-123.
Zhou Y, Philip AM, Chikovsky MN, et al. Implantation of XEN gel stent in a patient with ocular cicatricial pemphigoid. Am J Ophthalmol Case Rep. 2023;29:101801.
Soni A, Shah K, Shah M, Vazirani J. Peripheral Ulcerative Keratitis as a Manifestation of Drug-Induced Cicatrizing Conjunctivitis. Cureus. 2023;15(1):e34115. doi:10.7759/cureus.34115. PMID:36843735; PMCID:PMC9946815.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:21. doi:10.21037/aes-22-35.
Shanbhag SS, Nikpoor N, Rao Donthineni P, Singh V, Chodosh J, Basu S. Autologous limbal stem cell transplantation: a systematic review of clinical outcomes with different surgical techniques. The British journal of ophthalmology. 2020;104(2):247-253. doi:10.1136/bjophthalmol-2019-314081. PMID:31118185.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.