Das okuläre Schleimhautpemphigoid (ocular mucous membrane pemphigoid, OCP) ist eine chronisch fortschreitende autoimmune bullöse Erkrankung, die durch Autoantikörper gegen Adhäsionskomponenten der epithelialen Basalmembran, einschließlich der Konjunktiva, verursacht wird und zu einer fortschreitenden Vernarbung führt. Das Schleimhautpemphigoid (mucous membrane pemphigoid, MMP) ist eine Gruppe systemischer Erkrankungen, die mehrere Schleimhäute (Mund, Augenoberfläche, Nasopharynx, Larynx, Speiseröhre, äußere Genitalien usw.) befallen; diejenigen mit Augenveränderungen werden als okuläres Pemphigoid oder okuläres narbiges Pemphigoid (ocular cicatricial pemphigoid) bezeichnet. Fälle ohne Hautbeteiligung sind nicht selten.
In der Augenheilkunde wird OCP als eine repräsentative Erkrankung der narbigen Keratokonjunktivalepitheliopathie mit Erschöpfung der kornealen epithelialen Stammzellen angesehen und durchläuft eine stufenweise Progression von chronischer Konjunktivitis über Symblepharon bis hin zur kornealen Keratinisierung.
Diese Erkrankung ist selten. Eine Überwachungsstudie im Vereinigten Königreich berichtete eine jährliche Inzidenz der narbigen Konjunktivitis von etwa 1,3 pro 1 Million Einwohner, und die Prävalenz von OCP wird auf etwa 1 pro 10.000 bis 50.000 Personen geschätzt5). Sie tritt bevorzugt bei älteren Menschen im Alter von 60–80 Jahren auf, mit einem Frauen-zu-Männer-Verhältnis von etwa 2:15). Die Häufigkeit von Schleimhautläsionen ist im Mund am höchsten (etwa 85 %), gefolgt von den Augen (etwa 65 %); etwa 50 % der OCP-Patienten haben extraokuläre Läsionen wie im Mund oder auf der Haut2).
Sie beginnt schleichend als beidseitige chronische Konjunktivitis, und die Krankheitsdauer beträgt nicht selten mehrere Jahre bis über ein Jahrzehnt. Im Frühstadium treten nur unspezifische Konjunktivitis-Symptome auf, sodass viele Fälle lange Zeit als allergische Konjunktivitis, chronische Konjunktivitis oder trockenes Auge behandelt werden. Eine sorgfältige Anamnese und Beurteilung der Spaltlampenbefunde unter Berücksichtigung dieser Erkrankung sind für eine frühzeitige Diagnose und Verbesserung der Sehfunktionsprognose äußerst wichtig.
Überblick über die Schleimhautpemphigoid (MMP)
Zielorgane: Schleimhäute mit mehrschichtigem Plattenepithel im gesamten Körper, einschließlich Mundhöhle, Augenoberfläche, Nasopharynx, Larynx, Ösophagus und Genitalien
Hautläsionen: treten oft nicht begleitend auf. Erosionen und Narben stehen mehr im Vordergrund als Blasen
Systemische Komplikationen: Dysphagie durch Ösophagusstenose, Larynxstenose beeinflusst die Lebensprognose2)
Merkmale der okulären Pemphigoid (OCP)
Wesen der Augenläsionen: beidseitige chronische Konjunktivitis und fortschreitende konjunktivale Vernarbung
Ziel der Pathologie: Symblepharon, Erschöpfung der kornealen epithelialen Stammzellen, Keratinisierung der Augenoberfläche
Extraokuläre Läsionen: etwa 50 % der Fälle haben Beteiligung anderer Stellen wie Mundhöhle oder Haut2)
Schleichender Verlauf: kann lange Zeit asymptomatisch sein, einige Fälle werden erst bei Sehverschlechterung entdeckt2)
QSind Schleimhautpemphigoid (MMP) und okuläre Pemphigoid (OCP) dieselbe Erkrankung?
A
Schleimhautpemphigoid (MMP) ist ein Überbegriff für eine autoimmune bullöse Erkrankung, die mehrere Schleimhäute befällt, darunter Mundhöhle, Augen, Nasopharynx, Larynx, Ösophagus und äußere Genitalien. Davon werden diejenigen mit Augenläsionen als okuläre Pemphigoid (OCP) bezeichnet. OCP kann nur mit Augenläsionen oder mit extraokulären Läsionen wie Mundhöhle und Haut einhergehen; etwa 50 % der Fälle haben extraokuläre Läsionen2). Es ist nicht ungewöhnlich, dass Hautläsionen unauffällig sind, sodass der Augenarzt oft als erster die Diagnose stellt.
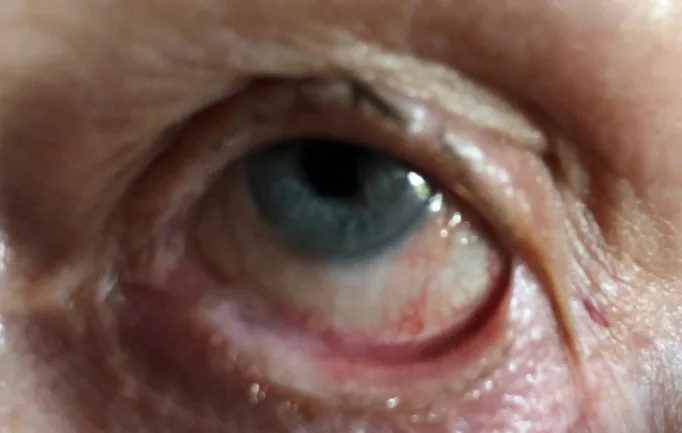
Stan C, Golea A, Gheorghe A, et al. Ocular cicatricial pemphigoid. Rom J Ophthalmol. 2020 Apr-Jun;64(2):226-230. Figure 2. PMCID: PMC7339695. License: CC BY.
Am linken Auge hat sich eine Symblepharon (Verklebung der Bindehaut von Lid und Augapfel) gebildet. Dies zeigt ein konkretes Beispiel einer Symblepharon, wie im Abschnitt über die Hauptsymptome und klinischen Befunde beschrieben.
Im Frühstadium stehen chronische Bindehautrötung, Trockenheitsgefühl, Fremdkörpergefühl, Brennen und Tränenfluss im Vordergrund. Diese sind resistent gegen die übliche Behandlung des trockenen Auges und geben bei längerem Bestehen Anlass, an diese Erkrankung zu denken 1). In fortgeschrittenen Fällen kommen Bewegungseinschränkungen des Auges durch Symblepharon, unvollständiger Lidschluss, Hornhautreizsymptome durch Trichiasis sowie Sehverschlechterung durch Hornhauttrübung oder konjunktivale Epitheleinwanderung hinzu.
Der Verlauf dieser Erkrankung ist schleichend; sie kann lange Zeit symptomlos bleiben und erst bei einer Sehverschlechterung entdeckt werden 2).
Zur Beurteilung des Fortschreitens wird die Foster-Klassifikation (modifizierte Tauber-Foster-Klassifikation) häufig verwendet. Sie beginnt mit einer chronischen Konjunktivitis im Stadium I, gefolgt von einer Verkürzung des unteren Fornix im Stadium II, Symblepharon und Hornhauteinwanderung von Blutgefäßen im Stadium III und schließlich einer Keratinisierung der Augenoberfläche im Stadium IV 2).
Verkürzung des unteren Fornix (A–D: Beurteilung des Fortschreitens in 25%-Schritten)
Stadium III
Symblepharon, Hornhauteinwanderung von Blutgefäßen, Trichiasis, verminderte Tränensekretion
Stadium IV
Keratinisierung der Augenoberfläche, Ankyloblepharon, schwere Trockenheit, hautartige Veränderung der Hornhaut
Anfangs werden auf der Lidbindehaut lineare oder fleckige weiße sehnenartige Narben beobachtet. Im fortgeschrittenen Stadium kommt es zu einer Verkürzung des Bindehautsacks, einem Verschwinden der Vogt’schen Hornhautlimbus-Palisaden (Palisades of Vogt, POV), einem Eindringen von Bindehautepithel in die Hornhaut, einer Verstopfung der Tränenkanäle, einem Verschwinden der Bindehaut-Becherzellen und einem schweren trockenen Auge durch das Verschwinden der Meibom-Drüsen. Bei akuten Exazerbationen kann ein ausgedehnter persistierender Epitheldefekt auftreten.
Histologisch zeigen sich eine Fibrose des Bindehautstromas, ein vollständiges Verschwinden der Becherzellen und eine Plattenepithelmetaplasie 1). Das Verschwinden der Becherzellen bedeutet einen Verlust der Muzinsekretionsfunktion der Bindehaut, wodurch die Stabilität des Tränenfilms erheblich beeinträchtigt wird. Zudem geht durch die Zerstörung der Meibom-Drüsen auch die Lipidschicht verloren, die Tränenverdunstung nimmt zu und es entsteht ein schweres verdunstungsbedingtes trockenes Auge.
Als systemische Schleimhautkomplikationen müssen Mundläsionen mit desquamativer Gingivitis oder bullösen Läsionen, Ösophagusläsionen mit Schluckbeschwerden sowie Kehlkopf- und Trachealläsionen mit Heiserkeit oder Atemnot beurteilt werden. Die Kehlkopfstenose ist ein lebensbestimmender Prognosefaktor, der eine Atemwegssicherung erfordert; bei Patienten mit Atemwegssymptomen ist eine dringende Zusammenarbeit mit der Hals-Nasen-Ohren-Heilkunde erforderlich 2). Die Ösophagusstenose verursacht Schluckbeschwerden und Mangelernährung; eine endoskopische Beurteilung durch einen Gastroenterologen und gegebenenfalls eine Ballondilatation werden in Betracht gezogen. Hautläsionen treten oft eher als narbige Erytheme oder atrophische Veränderungen auf, insbesondere an Kopf, Gesicht und Hals.
Das okuläre Schleimhautpemphigoid wird durch einen Autoimmunmechanismus ausgelöst (Einzelheiten zur Pathologie in Abschnitt 6). Zu den Risikofaktoren gehören höheres Alter, weibliches Geschlecht und eine Vorgeschichte anderer Autoimmunerkrankungen. Ein eindeutiger Auslöser wurde nicht identifiziert, aber es wird diskutiert, dass Infektionen oder Medikamentenexposition zum Zusammenbruch der Immuntoleranz beitragen könnten. Eine Assoziation mit HLA-DQB1*0301 wurde in ausländischen Studien berichtet, was auf einen genetischen Beitrag hindeutet.
Klinisch ist die Abgrenzung zu anderen Erkrankungen mit vernarbender Bindehautentzündung wichtig. Die unten aufgeführten Erkrankungen unterscheiden sich sowohl in der Pathologie als auch in der Behandlung, daher muss die Ursache bei der Erstvorstellung eingegrenzt werden.
Abzugrenzende Autoimmun- und entzündliche Erkrankungen
Stevens-Johnson-Syndrom / toxische epidermale Nekrolyse : akute mukokutane Schädigung durch Arzneimittelreaktion. Abgrenzung durch Fieber und generalisierten Hautausschlag in der Vorgeschichte
Graft-versus-Host-Krankheit (GVHD) : tritt nach hämatopoetischer Stammzelltransplantation auf. Bei schweren und fortschreitenden Verwachsungen ist ein gleichzeitiges Vorliegen einer okulären Schleimhautpemphigoid in Betracht zu ziehen 4)
Sarkoidose : nicht-verkäsende granulomatöse Bindehautentzündung, die vernarben kann. Wird als Nachahmer des okulären Schleimhautpemphigoids berichtet 3)
Medikamenteninduziertes Pseudo-Okularpemphigoid : Langzeitanwendung von Antiglaukomatosa (Latanoprost, Timolol, Brinzolamid etc.) ist ein typisches Beispiel. Besserung nach Absetzen des Medikaments 7)
Chemisches Trauma / Verbrennung : Alkaliverletzungen sind besonders schwerwiegend
Infektiös : Trachom ist in Entwicklungsländern eine Hauptursache. In Industrieländern schwere Fälle von adenoviraler Keratokonjunktivitis
Iatrogen : Zustand nach Augenoperation mit Bindehautschnitt
Der beidseitige, langsam fortschreitende chronische Entzündungsprozess des Hornhaut- und Bindehautepithels mit Narbenbildung ist ein starker Hinweis auf OCP. Insbesondere bei älteren Menschen, vor allem Frauen, mit Trichiasis oder Entropion sollte sorgfältig auf eine Verkürzung des Bindehautsacks oder Symblepharon geachtet werden.
Bei der Anamnese sollten Augenverletzungen, Augeninfektionen sowie die frühere und aktuelle Anwendung von Augentropfen und oralen Medikamenten detailliert erfragt werden. Darüber hinaus sollte eine systemische Überprüfung auf Schleimhautläsionen im Mund, Nasopharynx, Larynx, Ösophagus und der Haut erfolgen, und bei Bedarf sollte eine Zusammenarbeit mit den Fachabteilungen für Dermatologie, Hals-Nasen-Ohrenheilkunde, Gastroenterologie und Rheumatologie angestrebt werden2).
Impressionzytologie : Zytologische Untersuchung der Bindehaut zum Nachweis des Verlusts von Becherzellen
Fluorescein-Färbung : An Stellen, an denen das Bindehautepithel in die Hornhautoberfläche eindringt, wird eine erhöhte Durchlässigkeit für den Farbstoff beobachtet, was zusammen mit dem Verlust der POV ein Zeichen für eine Erschöpfung der Hornhautepithel-Stammzellen ist8)
Die definitive Diagnose stützt sich auf die Bindehautbiopsie. Eine Probe wird von der unteren Bulbusbindehaut entnommen, und es werden gleichzeitig eine histopathologische Untersuchung an formalinfixierten Schnitten und eine direkte Immunfluoreszenz (DIF) an in Michel-Lösung fixierten Schnitten durchgeführt5).
Der positive DIF-Befund bei OCP ist eine lineare Ablagerung von IgG, IgA, IgM und Komplement C3 entlang der Basalmembran des Bindehautepithels5). Allerdings sind falsch-negative DIF-Ergebnisse nicht selten. Ursachen sind die Ruhephase der Erkrankung, das vollständige Verschwinden der Basalmembran im Endstadium, eine ungeeignete Biopsiestelle und lokale Schwankungen der Immunreaktion. Bei klinisch starkem Verdacht auf OCP sollte die Erkrankung auch bei negativer DIF nicht ausgeschlossen werden, und durch mehrere Biopsien, einschließlich einer erneuten Biopsie des anderen Auges, kann eine verbesserte Sensitivität erwartet werden4,5).
Autoantikörper: Anti-BP180-Antikörper (Anti-Kollagen Typ XVII) und Anti-Laminin-332-Antikörper (Anti-Laminin-5) werden bei den meisten OCP-Patienten nachgewiesen. Diese Autoantikörper spielen eine zentrale Rolle in der Pathogenese.
Tests zur Differenzialdiagnose: BSG, Blutbild, Leber- und Nierenwerte, ANA, SS-A/SS-B (Sjögren-Syndrom), ANCA (Granulomatose mit Polyangiitis), ACE/Lysozym (Sarkoidose).
Auch bei normalem ACE/Lysozym bleibt Sarkoidose eine Differenzialdiagnose; negative Immunfluoreszenz hilft, OCP auszuschließen 3).
QWarum kann eine OCP nicht ausgeschlossen werden, wenn die direkte Immunfluoreszenz der Konjunktivalbiopsie negativ ist?
A
Der DIF-Test hat keine vollständige Sensitivität, und mehrere Faktoren können falsch-negative Ergebnisse verursachen. Hauptursachen sind die Ruhephase der Erkrankung, das vollständige Verschwinden der Basalmembran im Endstadium, die Entnahme der Probe aus einem wenig aktiven Bereich sowie die lokale Variabilität der Immunablagerungen. Es wurden auch Fälle mit unterschiedlichen Ergebnissen zwischen linkem und rechtem Auge berichtet 5). Wenn der klinische Befund stark auf eine OCP hindeutet, verbessert die Wiederholung der Untersuchung, einschließlich einer erneuten Biopsie vom anderen Auge oder einer anderen Stelle, die Sensitivität 4). Der Nachweis von Serum-Antikörpern gegen BP180 und Laminin 332 ist ebenfalls als diagnostische Hilfe nützlich.
Die Behandlungsziele sind das Stoppen der fortschreitenden Vernarbung, die Prävention und Korrektur von Hornhaut- und Lidkomplikationen sowie die Symptomlinderung. Diese Erkrankung ist eine systemische Autoimmunerkrankung; eine alleinige Lokaltherapie kann das Fortschreiten nicht kontrollieren. Die Hauptbehandlung ist die systemische Immunsuppression, und eine Zusammenarbeit mit der Dermatologie und Rheumatologie wird dringend empfohlen 2).
Die Medikamente werden je nach Krankheitsaktivität und Entzündungsaktivität stufenweise ausgewählt. In allen Fällen sind eine Überwachung der Nebenwirkungen und regelmäßige Blutuntersuchungen unerlässlich 2,5).
Systemische Therapie: Stufenweise Auswahl
Leicht bis mittelschwer: Dapson (Diaminodiphenylsulfon) 50–200 mg/Tag als erste Wahl. Ein Ausschluss eines G6PD-Mangels und die Überwachung auf Anämie und Methämoglobinämie sind obligatorisch 5)
Dapson unwirksam/mittelschwer: Umstellung auf Mycophenolatmofetil 1–3 g/Tag, Azathioprin 1–2 mg/kg/Tag oder Methotrexat 7,5–25 mg/Woche 2)
Schwer/akuter Schub: Kombination von Cyclophosphamid 1–2 mg/kg/Tag und Prednisolon in kurzer hochdosierter Therapie 5)
Symptomatische Therapie: Niedrig dosierte Steroid-Augentropfen zur Entzündungshemmung, konservierungsmittelfreie künstliche Tränen zur Behandlung des trockenen Auges, Epilation der Wimpern zur Reduzierung der Hornhautreizung
Rekonstruktion von Symblephara: Nach Beruhigung der Entzündung Lösung der Verwachsungen und Rekonstruktion mittels Amnionmembran- oder Mundschleimhauttransplantation 2)
Fälle mit Glaukom: Umstellung auf konservierungsmittelfreie Augentropfen als Grundlage. Es gibt auch Fallberichte über minimalinvasive Glaukomchirurgie (XEN-Gel-Stent usw.) 6)
Endstadium: Transplantation von kultiviertem Schleimhautepithel zur Rekonstruktion des Hornhautepithels, Hornhautprothese (Boston KPro Typ 2, OOKP) bei schweren Fällen
Die meisten Medikamente sind für diese Erkrankung nicht von der Krankenkasse erstattungsfähig, und vor der Einleitung ist eine ausführliche Aufklärung des Patienten sowie ethische Abwägung erforderlich. Es ist zu beachten, dass Ciclosporin und Cyclophosphamid oral ebenfalls nicht für diese Indikation zugelassen sind. Dapson kann Hämolyse der roten Blutkörperchen und Methämoglobinämie auslösen; daher sollte vor der Verabreichung die Aktivität der Glucose-6-Phosphat-Dehydrogenase (G6PD) gemessen werden, um einen Mangel auszuschließen. Nach Behandlungsbeginn sind regelmäßige Blutbild-, Leber- und Nierenfunktionskontrollen sowie eine schrittweise Dosissteigerung erforderlich. Mycophenolatmofetil ist relativ gut verträglich und für die Langzeitanwendung geeignet, jedoch sollte ein Screening auf opportunistische Infektionen unter Berücksichtigung des Risikos erfolgen. Cyclophosphamid ist ein starkes Immunsuppressivum für schwere Fälle, birgt jedoch Risiken wie Myelosuppression, hämorrhagische Zystitis und langfristig ein Krebsrisiko; daher muss die Gesamtdosis überwacht werden. Rituximab, ein monoklonaler Anti-CD20-Antikörper, wird als Rescue-Therapie bei refraktärer OCP eingesetzt; eine Risikobewertung der Hepatitis-B-Virus-Reaktivierung vor der Verabreichung ist wichtig2).
Die lokale Behandlung ist als begleitende symptomatische Therapie zur systemischen Behandlung wichtig. Niedrig dosierte Steroid-Augentropfen lindern die Entzündung der Augenoberfläche, und konservierungsmittelfreie künstliche Tränen behandeln das trockene Auge. Chirurgische Eingriffe wie Punctum-Plugs sollten aufgrund des Risikos einer beschleunigten Narbenbildung sorgfältig abgewogen werden.
Chirurgische Behandlung und perioperatives Management
Eine chirurgische Behandlung sollte nur bei vollständig kontrollierter Entzündung durchgeführt werden. Eine Operation unter aktiver Entzündung kann zu einer raschen Progression von Narbenbildung, Adhäsionen und Keratinisierung führen2).
Kataraktoperation: Falls erforderlich, wird sie nach ausreichender Entzündungshemmung durch systemische Immunsuppression am Operationstag durchgeführt. Bei unzureichender Entzündungshemmung können postoperativ Adhäsionen und Keratinisierung rasch fortschreiten.
Rekonstruktion des Hornhautepithels: Bei persistierenden Epitheldefekten aufgrund einer akuten Exazerbation wird zunächst eine konservative Behandlung mit oralen Steroiden durchgeführt. Bei refraktären Fällen sind die Transplantation von autologen oder allogenen kultivierten Hornhautepithelblättern oder die autologe Mundschleimhautepitheltransplantation Optionen; die autologe Mundschleimhautepitheltransplantation ist in Japan als fortgeschrittene medizinische Behandlung anerkannt.
Operation zur Sehverbesserung im Endstadium: Bei schweren Fällen mit konjunktivaler Invasion und Keratinisierung der Hornhaut hat eine chirurgische Behandlung zur Sehverbesserung eine schlechte Prognose und wird in der Regel nicht durchgeführt.
Bei beidseitigen fortschreitenden Erkrankungen wie schwerer OCP sind die Indikationen für eine autologe Limbusstammzelltransplantation (CLAu, CLET, SLET) begrenzt. Viele beidseitige Fälle haben kein gesundes Spenderauge, und eine allogene Transplantation oder eine Transplantation von kultiviertem Mundschleimhautepithel wird in Betracht gezogen 9). Eine systematische Übersicht zur autologen Limbusstammzelltransplantation, hauptsächlich bei einseitigen chemischen Verletzungen, berichtete über eine anatomische Erfolgsrate von 69 % und eine funktionelle Erfolgsrate von 60 % bei 1.023 Augen, aber sie ist bei beidseitigen Autoimmunerkrankungen wie OCP nicht die erste Wahl 9).
QWie kann eine Kataraktoperation bei OCP-Patienten sicher durchgeführt werden?
A
Eine Operation bei aktiver Entzündung ist ein wesentlicher Risikofaktor für die rasche Progression von Symblepharon und Hornhautverhornung. Bei der Planung einer Operation ist es notwendig, ab dem Operationstag eine systemische immunsuppressive Therapie (Steroide, Ciclosporin, Cyclophosphamid usw.) zu kombinieren, um die Entzündung vollständig zu beruhigen, die physische Schädigung der Augenoberfläche während der Operation zu minimieren und die Immunsuppression auch nach der Operation fortzusetzen. Wenn die Operation bei unzureichender Entzündungskontrolle vor der Operation durchgeführt wird, besteht ein hohes Risiko für eine rasche Progression von Verwachsungen und Verhornung nach der Operation. Die Wahl des Operationsverfahrens, das systemische Management und die Zusammenarbeit mit der Dermatologie und Rheumatologie sind äußerst wichtig 2).
6. Pathophysiologie und detaillierter Krankheitsmechanismus
OCP ist eine Typ-II-Autoimmunerkrankung (zytotoxisch), die durch die Produktion von Autoantikörpern gegen Hemidesmosom-assoziierte Basalmembrankomponenten gekennzeichnet ist 5).
BP180 (Kollagen Typ XVII) : Ein Transmembranprotein der Hemidesmosomen, das epidermale/epitheliale Zellen mit der Basalmembran verbindet. Anti-BP180-Antikörper sind einer der am häufigsten nachgewiesenen Autoantikörper bei OCP-Patienten.
Laminin 332 (Laminin 5) : Ein Adhäsionsprotein in der Basalmembran, das Hemidesmosomen mit Kollagen Typ IV verbindet. Anti-Laminin-332-Antikörper werden ebenfalls bei vielen OCP-Patienten nachgewiesen.
Weitere Antikörper gegen α6β4-Integrin und andere Basalmembrankomponenten wurden ebenfalls berichtet.
Wenn Autoantikörper an die Basalmembran des Bindehautepithels binden, wird das Komplementsystem aktiviert und Entzündungszellen werden rekrutiert. In der akuten Phase sind Eosinophile und Neutrophile die Hauptakteure der Entzündung, während in der chronischen Phase die lymphozytäre Infiltration überwiegt 5). Die freigesetzten Zytokine und Proteasen aktivieren Fibroblasten unter dem Bindehautepithel und fördern eine übermäßige Produktion extrazellulärer Matrix wie Kollagen, was zu Fibrose und Narbenbildung der Bindehaut führt. Th2-Zytokine wie Transforming Growth Factor β (TGF-β), Interleukin-4 (IL-4), IL-5 und IL-13 sind in den Läsionen hochreguliert und gelten als Haupttreiber der Fibrose. Ein Ungleichgewicht zwischen Matrix-Metalloproteinase (MMP-9) und Gewebeinhibitor (TIMP) führt zu einem unkontrollierten Remodeling des Bindehautgewebes, was ebenfalls ein pathologisches Merkmal ist.
Erschöpfung der kornealen epithelialen Stammzellen und Keratinisierung der Hornhaut
Das schrittweise Fortschreiten der Bindehautnarbenbildung entspricht der Foster-Klassifikation. Anhaltende chronische Entzündung führt zu subkonjunktivaler Fibrose (Stadium I), Verkürzung des Fornix (Stadium II), Symblepharon (Stadium III) und schließlich zum Verschwinden der Palisaden von Vogt (POV) des Hornhautlimbus8). Die POV enthalten korneale epitheliale Stammzellen, daher bedeutet ihr Verlust das Versagen der normalen Regeneration des Hornhautepithels. Die Erschöpfung der kornealen epithelialen Stammzellen führt zu einer „Konjunktivalisierung“, bei der das Bindehautepithel die Hornhautoberfläche invadiert, was zu Hornhauttrübung, Gefäßeinsprossung und schließlich zu einer hautfähnlichen Keratinisierung (Stadium IV) führt 8).
Die chronische okuläre Graft-versus-Host-Krankheit (oGVHD) nach hämatopoetischer Stammzelltransplantation zeigt sich typischerweise mit chronischer Entzündung der Augenoberfläche, trockenem Auge und leichter Bindehautfibrose. Bei schwerer und schnell fortschreitender Narbenbildung mit Symblepharon sollte jedoch eine Koexistenz mit OCP in Betracht gezogen werden 4). oGVHD und OCP haben ähnliche klinische Erscheinungsbilder, aber unterschiedliche Behandlungsstrategien, daher ist eine Unterscheidung mittels Bindehautbiopsie und DIF wichtig 4).
QWas ist die Erschöpfung der kornealen epithelialen Stammzellen bei OCP?
A
Der Hornhautlimbus enthält Stammzellen, die für die Regeneration des Hornhautepithels verantwortlich sind und als Palisaden von Vogt (POV) sichtbar sind. Bei OCP erstrecken sich chronische Entzündung und Narbenbildung auf den Limbus, was zum Verlust der POV und der Stammzellen führt. Die Erschöpfung der Stammzellen verursacht eine „Konjunktivalisierung“, bei der das Bindehautepithel die Hornhautoberfläche invadiert, wodurch eine normale Regeneration des Hornhautepithels unmöglich wird. Dies führt zu Hornhauttrübung, Gefäßeinsprossung und Keratinisierung, was zu schwerer Sehbehinderung führt 8). Dieser Zustand ist das Endstadium schwerer Augenoberflächenerkrankungen, die bei okulärem Schleimhautpemphigoid, Stevens-Johnson-Syndrom und chemischen Traumata gemeinsam auftreten.
Die Wirksamkeit von Rituximab und IVIG bei refraktärer OCP wurde in Fallserien gezeigt, und sie etablieren sich als Behandlungsoptionen für Fälle, die auf konventionelle Immunsuppression resistent sind 2). Die Verabreichung von Rituximab eliminiert Autoantikörper-produzierende B-Zellen, und es wurden Fälle mit klinischer Remission berichtet. IVIG vereint entzündungshemmende und immunmodulatorische Wirkungen und wird als Begleittherapie bei schweren oder therapieresistenten Fällen eingesetzt. Das Potenzial neuer Immunmodulatoren wie JAK-Inhibitoren wird diskutiert, und ihre orale Verabreichung erleichtert die ambulante Behandlung, was zukünftige klinische Anwendungen erwarten lässt.
Im Bereich der Rekonstruktion des Hornhautepithels werden transplantierte kultivierte Hornhautepithelblätter und autologe Mundschleimhautepithelblätter klinisch eingesetzt, und ihre Entwicklung als fortgeschrittene Medizin schreitet voran. Autologe Hornhautepithelblätter werden aus Hornhautepithelzellen des gesunden Auges hergestellt, können aber bei bilateraler OCP nicht verwendet werden; daher werden allogene Hornhautepithelblätter aus Hornhäuten von Augenbanken oder autologe Mundschleimhautepithelblätter aus den eigenen Mundschleimhautzellen des Patienten gewählt. Autologe Mundschleimhaut birgt kein Abstoßungsrisiko und ermöglicht eine langfristige Stabilisierung der Augenoberfläche. Trägermaterialien wie Amnion, Fibrinkleber, temperaturresponsive Kulturplatten und Polyvinylidenfluorid (PVDF)-Membranen wurden entwickelt und erweitern die chirurgischen Optionen 8).
Bei Glaukomfällen mit Vernarbung der Bindehaut, die eine konventionelle filtrierende Operation erschwert, wurden erfolgreiche Fälle einer minimalinvasiven Glaukomchirurgie (MIGS) mit dem XEN-Gel-Stent berichtet. Nach Kontrolle der Augenoberflächenentzündung wird der Stent mittels ab-interno-Technik eingesetzt, und es wurden Fälle mit Augeninnendruckkontrolle ohne Augentropfen und Entzündungsberuhigung ein Jahr postoperativ gezeigt, was als neuer Ansatz für Glaukom bei schweren Augenoberflächenerkrankungen Beachtung findet 6). Zukünftig werden multizentrische Studien zur Sammlung von Langzeitergebnissen und die Bewertung des Therapieansprechens mittels Biomarkern in diesem Bereich erwartet.
Farrag A, Chan A, Tong L. Cicatricial Conjunctivitis and Concurrent Clinical Features: A Case Study. Clinical medicine insights. Case reports. 2022;15:11795476221100605. doi:10.1177/11795476221100605. PMID:35601266; PMCID:PMC9121463.
Razzak A, Ait Ammar H, Bouazza M, Elbelhadji M. Accidental Discovery of Ocular Cicatricial Pemphigoid. Cureus. 2025;17(1):e77425. doi:10.7759/cureus.77425. PMID:39949452; PMCID:PMC11823481.
Murati Calderon RA, López-Fontanet JJ, Ramirez Marquez E, Tavares Reigada AS, Oliver A. Sarcoidosis: A Mimicker of Ocular Cicatricial Pemphigoid. Cureus. 2025;17(10):e93862. doi:10.7759/cureus.93862. PMID:41194826; PMCID:PMC12584608.
Taketani Y, Dehghani S, Sinha S, Freitag SK, Papaliodis G, Foster S, et al. Concurrence of Ocular Cicatricial Pemphigoid in Chronic Ocular Graft-Versus-Host Disease. Cornea. 2024;43(3):387-390. doi:10.1097/ICO.0000000000003386. PMID:38128104; PMCID:PMC12703942.
Tesorero JCC, Sosuan GMN, Lim Bon Siong R. Ocular Cicatricial Pemphigoid in a Healthy Elderly Male Filipino Patient. Acta Med Philipp. 2025;59(18):117-123.
Zhou Y, Philip AM, Chikovsky MN, et al. Implantation of XEN gel stent in a patient with ocular cicatricial pemphigoid. Am J Ophthalmol Case Rep. 2023;29:101801.
Soni A, Shah K, Shah M, Vazirani J. Peripheral Ulcerative Keratitis as a Manifestation of Drug-Induced Cicatrizing Conjunctivitis. Cureus. 2023;15(1):e34115. doi:10.7759/cureus.34115. PMID:36843735; PMCID:PMC9946815.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:21. doi:10.21037/aes-22-35.
Shanbhag SS, Nikpoor N, Rao Donthineni P, Singh V, Chodosh J, Basu S. Autologous limbal stem cell transplantation: a systematic review of clinical outcomes with different surgical techniques. The British journal of ophthalmology. 2020;104(2):247-253. doi:10.1136/bjophthalmol-2019-314081. PMID:31118185.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.