血液檢查

動脈炎性前部缺血性視神經病變(AAION)
一目瞭然的要點
Section titled “一目瞭然的要點”1. 什麼是動脈炎性前部缺血性視神經病變(AAION)?
Section titled “1. 什麼是動脈炎性前部缺血性視神經病變(AAION)?”動脈炎性前部缺血性視神經病變(AAION)是由於營養血管的血管炎導致視神經缺血所致。血管炎導致血管壁增厚、管腔狹窄和血栓形成,引起缺血性壞死。供應視盤的睫狀後短動脈(SPCAs)的血管炎被認為是其本質。AAION佔所有前部缺血性視神經病變的5-10%,大多數為非動脈炎性前部缺血性視神經病變(NAION)。
最常見的原發病是巨細胞動脈炎(GCA,舊稱顳動脈炎)。其他原因包括帶狀皰疹、復發性多軟骨炎、高安動脈炎、類風濕關節炎、結節性多動脈炎、系統性紅斑狼瘡和過敏性肉芽腫性血管炎(Churg-Strauss症候群)。
巨細胞動脈炎(GCA)的概述與歷史
Section titled “巨細胞動脈炎(GCA)的概述與歷史”GCA是一種累及中大型動脈的全身性肉芽腫性血管炎。最早的描述可追溯到10世紀巴格達的Ali Ibn Isa al-Kahhal。1890年,Hutchinson描述了伴有頭痛的紅色條紋;1932年,Bayard Horton進行了首次顳動脈活檢,將其描述為肉芽腫性血管炎。1941年,Gilmour首次描述了巨細胞,從而確立了現在的病名。
多見於50歲以上女性(男女比1:3),70歲以上發生率急遽增加。GCA發病中位年齡為75歲。AAION的估計年發生率在50歲以上族群為每10萬人0.36例。
GCA的視覺併發症發生率為10-30%(有報告高達70%),AAION占GCA相關視力喪失的60-90% 3)。GCA發生率隨年齡增加而上升,60歲世代為每10萬人2.3例,90歲世代達44.7例。
北歐白人最常見(挪威約每10萬人30例),黑人和東方人罕見。日本發生率為每10萬人1.47例,遠低於歐美。在歐洲,它是50歲以上人群中最常見的原發性全身性血管炎,每年每百萬人中有32-290例發病 6)。
Q
AAION與非動脈炎性前部缺血性視神經病變(NAION)有何不同?
2. 主要症狀和臨床所見
Section titled “2. 主要症狀和臨床所見”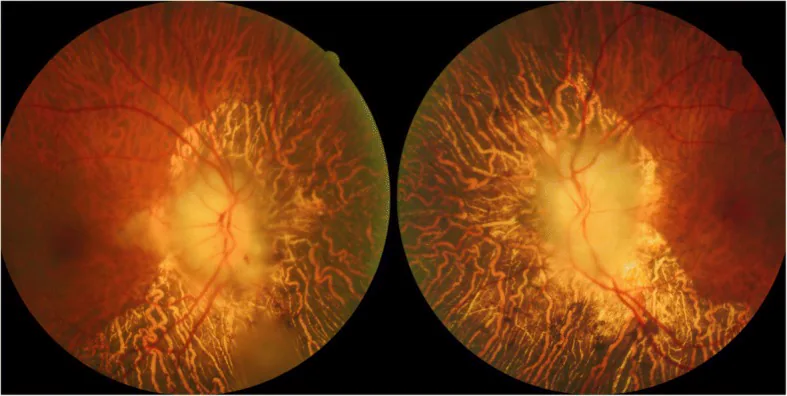
Tian G, et al. Giant cell arteritis presenting as bilateral anterior ischemic optic neuropathy: a biopsy-proven case report in Chinese patient. BMC Ophthalmol. 2018. Figure 1. PMCID: PMC6208180. License: CC BY.
初診時的眼底照片,顯示嚴重的雙側視盤水腫,呈粉筆樣蒼白外觀,右眼有線狀出血和軟性滲出,以及瀰漫性視盤周圍脈絡膜萎縮。這與本文「2. 主要症狀和臨床所見」一節中討論的「粉筆樣蒼白水腫」相對應。
GCA在老年人中表現為急性單眼或雙眼視力下降。許多患者有短暫性黑矇作為前驅症狀。若不治療,短期內對側眼常受累。
- 急性視力下降:超過60%出現嚴重障礙,視力低於20/200。超過20%進展為無光感,導致嚴重視功能損害。
- 短暫性黑矇:約30%的永久性視力喪失病例出現前驅症狀,平均在發病前8.5天出現。報告發生率為2-19%。在NAION中極為罕見,是重要的鑑別點。
- 頭痛:最常見的全身症狀,發生率為65-90%。新發的顳部或枕部頭痛具有特徵性 1)。
- 下頷跛行:咀嚼時下頷疼痛或疲勞。是GCA最特異的症狀。發生率為11-45% 1)。
- 頭皮壓痛:顳動脈或頭皮區域的壓痛。梳頭或枕在枕頭上時出現不適。
- 全身症狀:發燒、體重減輕、倦怠感、食慾不振、肌肉痛、關節痛等。
- PMR(風濕性多發性肌痛症)症狀:高達50%合併出現。表現為雙側頸部、肩部、骨盆的疼痛和僵硬。
- 複視:由第3、4、6腦神經麻痺引起。10~15%發生3)。
- 眼球運動限制:可能由腦神經麻痺引起。
- 潛在性GCA(occult GCA):高達20%的AAION患者缺乏明顯的全身症狀。
Q
沒有全身症狀也可能有AAION嗎?
A
有可能。稱為潛在性巨細胞動脈炎(occult GCA)的病症存在於高達20%的AAION患者,缺乏頭痛、顎跛行等典型全身症狀。不能因為沒有全身症狀就否定巨細胞動脈炎,必須透過血液檢查(ESR、CRP)和顳動脈切片進行評估。
臨床所見(醫師檢查確認的所見)
Section titled “臨床所見(醫師檢查確認的所見)”- 蒼白視乳頭水腫(pallid swelling):AAION的典型所見。表現為白堊色蒼白水腫(chalky-white pallor),與NAION的充血性水腫形成對比。
- 火焰狀出血:可見於視乳頭周圍。
- 軟性白斑(cotton wool spots):可見於後極部。
- 視乳頭周圍視網膜小動脈變細。
- 睫狀視網膜動脈阻塞:AAION相對特異的所見。
- 視網膜中央動脈阻塞(CRAO):也可能合併出現。
- RAPD陽性:單眼性或不對稱性視神經病變時相對性傳入瞳孔缺陷(RAPD)呈陽性。
- 視野缺損:水平半盲(altitudinal field defect)最為常見。
- 對側眼視神經盤:正常(與NAION的disc at risk不同)。
- 視神經萎縮與視神經盤凹陷:發病後6-8週出現視神經萎縮並伴隨視神經盤凹陷。90%以上的AAION病例可見3)。
- 顳動脈異常:怒張、結節、壓痛、搏動減弱或消失。
- 霍納氏症候群:罕見合併,表現為部分眼瞼下垂與瞳孔縮小5)。
- 螢光眼底攝影:視神經盤充盈延遲、視神經盤周圍脈絡膜充盈延遲或缺損(節段性缺血)具有特徵性。
3. 原因與風險因素
Section titled “3. 原因與風險因素”發病機制概述
Section titled “發病機制概述”在血管炎中,血管壁增厚導致管腔狹窄和血栓形成,引起缺血性壞死。短後睫狀動脈(SPCAs)的血管炎導致前部視神經缺血,並伴有節段性脈絡膜缺血。
眼部的直接機制如下:
- 短後睫狀動脈發炎→血管壁增厚→管腔狹窄→血栓形成→視神經盤缺血。
- 內側SPCA阻塞最為常見(20%的病例特別受影響)。
- SPCA供應篩板前區和篩板區,也參與視神經盤周圍脈絡膜循環。
GCA的血管炎機制
Section titled “GCA的血管炎機制”血管壁中的樹突狀細胞是疾病的主要貢獻因素。巨噬細胞和T細胞通過外膜的滋養血管(vasa vasorum)侵入,啟動致病級聯反應,導致影響中大型動脈的肉芽腫性血管炎。
主要風險因素
Section titled “主要風險因素”- 年齡:最大的風險因素。中位年齡75歲。50歲以下發病極為罕見。
- 性別:女性風險高2~6倍。
- 種族:北歐白種人最常見。黑人和亞洲人罕見。
- 遺傳因素:HLA-DRB1*04、DRW6和DR3與易感性增加相關。TNF-α基因座和IL-10啟動子的多態性也與風險增加相關。非白種人中也有家族性GCA的報導7)。
- 環境和感染因素:水痘-帶狀皰疹病毒(VZV)、肺炎衣原體和細小病毒B19的參與已被提出。
- 年齡相關機制:內彈性膜、彈性蛋白和細胞外基質的鈣化可能解釋了年齡特異性表現。
- 吸菸、低BMI、早停經:均被報導為風險因素。
- PMR合併:GCA和PMR顯示出強關聯。
- COVID-19:有報導稱疫情期間GCA發病率增加了70%2)。SARS-CoV-2對血管內皮有親和力,與血管炎的相似性提示了病理聯繫。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”AAION的診斷與巨細胞動脈炎的明確診斷並行進行。由於AAION和NAION的鑑別直接影響治療策略,快速系統的評估至關重要。
AAION與NAION的鑑別
Section titled “AAION與NAION的鑑別”| 項目 | AAION | NAION |
|---|---|---|
| 年齡 | 50歲以上(多為75歲以上) | 40歲以上 |
| 視力下降程度 | 重度(多低於20/200) | 相對較輕 |
| 視盤表現 | 蒼白水腫 | 充血性水腫(發紅腫脹) |
| 對側眼視盤 | 正常(非危險視盤) | 危險視盤(小視盤、小凹陷) |
| 全身症狀 | 有(發燒、頭痛、下巴跛行) | 無 |
| 發炎標記 | ESR/CRP升高 | 正常 |
| 暫時性黑矇 | 常先發生 | 極罕見 |
| 脈絡膜充盈延遲 | 有(節段狀) | 不具特徵性 |
| 治療 | 必須大劑量類固醇 | 無確立治療 |
顳動脈活檢(TAB)
定位:巨細胞動脈炎確診的金標準。操作得當的情況下,敏感性和特異性均超過95%。
陽性發現:內膜增厚、內彈性膜斷裂、伴有巨細胞的慢性炎性浸潤。病理確診需要內彈性膜破壞和炎性細胞浸潤(急性期)或纖維化(慢性期)。巨細胞並非確診所必需。
假陰性:由於跳躍性病變,假陰性率為3–5%。有報導稱最高可達61%6)。即使TAB陰性也不能排除GCA。
實施時機:即使在開始類固醇治療後,也應在數天內進行活檢。
影像學檢查
顳動脈超音波(CDUS):無創、可重複。敏感性77%,特異性96%4)。特徵性表現包括暈徵(血管壁增厚導致的低迴聲環)、壓迫徵、狹窄和閉塞。由於跳躍性病變,雙側和多區域的全面檢查很重要4)。雙側暈徵陽性時,特異性升至100%4)。
PET-CT:可檢測大血管型GCA(LV-GCA)中主動脈及其分支的異常攝取。GAPS研究中敏感性92%,特異性85%6)。
MRI:有助於鑑別AAION和NAION。評估視神經鞘和眼眶脂肪的對比增強(中央亮點)。
眼科檢查
ACR分類標準(1990年)及2022年ACR/EULAR分類標準
Section titled “ACR分類標準(1990年)及2022年ACR/EULAR分類標準”1990年ACR分類標準要求滿足以下5項中的至少3項。
| 項目 | 標準 |
|---|---|
| 發病年齡 | ≥50歲 |
| 新發頭痛 | 新出現的侷限性頭痛 |
| 顳動脈異常 | 壓痛或搏動減弱 |
| 紅血球沉降速率 | ≥50 mm/h |
| 動脈切片 | 單核細胞浸潤或肉芽腫性發炎 |
2022年ACR/EULAR分類標準新增了CRP≥10 mg/L,使診斷更全面4)。
需與結節性多動脈炎、肉芽腫性多血管炎(韋格納肉芽腫)、SLE等其他血管炎鑑別。GCA不侵犯肺和腎臟是重要的鑑別點。眼梅毒可能出現類似GCA的症狀10)。也需注意無全身症狀的潛在性GCA(約20%)。
Q
顳動脈切片陰性也不能排除巨細胞動脈炎嗎?
A
不能排除。由於跳躍性病變(發炎僅存在於血管的一部分),偽陰性率為3–5%(有報告指出最高可達61%6))。即使TAB陰性,若臨床高度懷疑GCA且ESR、CRP升高,應繼續治療。綜合評估臨床表現、血液檢查和超音波檢查結果非常重要。
5. 標準治療方法
Section titled “5. 標準治療方法”一旦懷疑視力障礙,應立即開始治療,無需等待切片確診。治療的主要目的是預防對側眼發病,患眼視力改善的可能性很小。僅15–20%的患者治療後視力改善。建議住院進行大劑量類固醇靜脈滴注治療。
類固醇治療(急性期、維持期、減量)
Section titled “類固醇治療(急性期、維持期、減量)”- 急性期:立即靜脈注射甲基潑尼松龍1g/日,持續3–5天。
- 維持期:改為口服潑尼松龍1mg/kg/日。
- 減量:根據全身狀況和血沉,至少4–6個月緩慢減量。部分病例可能需要1年以上。
- 注意:不推薦隔日給藥。
若無眼部和中樞神經系統症狀,可從潑尼松龍30–40mg/日開始治療。
減量方案(參考)
Section titled “減量方案(參考)”| 劑量 | 持續時間 |
|---|---|
| 普賴松 60 mg | 2週 |
| 普賴松 50 mg | 2週 |
| 普賴松 40 mg | 2週 |
| 普賴松 30 mg | 1週 |
| 普賴松 20 mg | 1週 |
| 普賴松 10 mg | 1週 |
根據全身狀況、ESR和CRP個別調整減量速度。
類固醇節約藥物(托珠單抗、甲氨蝶呤)
Section titled “類固醇節約藥物(托珠單抗、甲氨蝶呤)”當長期使用類固醇引起的副作用(庫欣樣症候群、高血糖、骨質疏鬆、消化道症狀等,約60%發生)成為問題時考慮使用。
- 托珠單抗(tocilizumab;IL-6受體抑制劑):2017年獲FDA核准用於GCA治療。隨機對照試驗(RCT)證明其具有糖皮質激素節省效應,並在12個月內達到緩解的有效性4)。有報告指出對類固醇抗性AAION有效。在COVID-19疫苗接種後的GCA病例中,也有使用TCZ 162mg皮下注射的報告8)9)。
- 甲氨蝶呤:提高糖皮質激素持續停用率,降低復發風險。病例報告中採用15mg/週的合併用藥1)。復發時,有時建議增加糖皮質激素劑量並合併MTX。
- 低劑量阿斯匹靈:可考慮用於預防心臟和腦血管缺血性併發症。
Q
糖皮質激素治療能否恢復視力?
A
患眼視力改善幾乎不可預期。視力改善僅約15-20%,多數病例殘留視力下降。糖皮質激素治療的主要目的是預防對側眼發病。
6. 病理生理學與詳細發病機轉
Section titled “6. 病理生理學與詳細發病機轉”巨細胞動脈炎的病理涉及兩種主要免疫反應機轉。它是一種T細胞媒介的肉芽腫性血管炎,起始於血管壁樹突細胞的活化,選擇性侵犯中至大型動脈。
免疫與炎症級聯反應(雙軸模型)
Section titled “免疫與炎症級聯反應(雙軸模型)”全身性炎症反應
抗原特異性反應
侵入動脈壁:巨噬細胞和T細胞利用外膜滋養血管(vasa vasorum)侵入。
免疫級聯反應:活化T細胞→CD4+ T細胞募集→Th1/Th17極化→IFN-γ/IL-17產生→單核細胞募集→巨噬細胞分化→巨細胞形成。
血管壁破壞:金屬蛋白酶和活性氧中間體導致內彈性膜破壞。
B細胞不參與:尚未證實B細胞參與,這是與ANCA相關血管炎的重要鑑別點。
視神經的血液供應與缺血
Section titled “視神經的血液供應與缺血”- 視神經的血液供應主要來自睫狀後短動脈(SPCA)和視網膜中央動脈的分支。
- SPCA供應篩板前區和篩板區,並參與視乳頭周圍脈絡膜循環。
- 在巨細胞動脈炎(GCA)中,SPCA的血栓性閉塞(20%病例特別受累)導致視盤缺血。
- 急性前部缺血性視神經病變(AAION)的屍檢顯示視盤水腫,伴有篩板前、篩板和篩板後區壞死及慢性炎症細胞浸潤。
- 螢光素眼底血管攝影數據支持SPCA受累的組織病理學證據。
內彈性膜斷裂是特徵性表現,巨細胞位於斷裂的內彈性膜附近。急性期以淋巴球浸潤為主,慢性期發生纖維化。作為對發炎的癒合反應,出現內膜增厚、肌纖維母細胞增殖和細胞外基質沉積,導致血管狹窄和阻塞。
7. 最新研究與未來展望(研究階段報告)
Section titled “7. 最新研究與未來展望(研究階段報告)”烏帕替尼(JAK1抑制劑)
Section titled “烏帕替尼(JAK1抑制劑)”口服JAK1選擇性抑制劑烏帕替尼於2025年獲得FDA批准用於治療GCA。作為靶向IL-6–JAK–STAT路徑的新型治療選擇而備受關注。
托珠單抗的RCT證據
Section titled “托珠單抗的RCT證據”托珠單抗在RCT中證明了類固醇節省效應和12個月內達到緩解的有效性4),並正在成為減少長期使用類固醇相關毒性的替代療法。
快速通道(超音波快速診斷路徑)
Section titled “快速通道(超音波快速診斷路徑)”利用超音波的巨細胞動脈炎快速診斷路徑正在歐洲普及。研究表明,引入快速通道可減少視力喪失、抑制過度治療並改善成本效益4)。超音波無創、可重複,並能同時評估多個動脈區域,因此被定位為早期診斷的主要工具。
GCA-MDS亞型與低甲基化藥物
Section titled “GCA-MDS亞型與低甲基化藥物”在骨髓增生異常症候群(MDS)背景下發生的GCA亞型(GCA-MDS)已被認識。據報導,10-20%的MDS患者會出現自體免疫疾病。GCA-MDS的典型症狀(頭痛、下頷跛行、AAION等)盛行率可能低於典型GCA,易出現類固醇依賴,無類固醇存活率和無復發存活率較低。添加低甲基化藥物(阿扎胞苷/地西他濱)可能有益,一項前瞻性研究(NCT02985190)正在進行中。最大規模的報告是2019年法國多中心21例。
COVID-19與GCA的關聯
Section titled “COVID-19與GCA的關聯”有報告稱,COVID-19大流行期間GCA發生率增加了70%2)。多項報告顯示,2020年GCA病例增加,眼部併發症率上升,推測與內皮損傷、Th1免疫和單核巨噬細胞系統有關2)。也有病例報告提示SARS-CoV-2可能觸發了GCA。
已有數個COVID-19疫苗接種後發生GCA的病例報告。Yoshimoto等人(2023)回顧了14例病例,報告從發病到診斷的時間為2週至4個月(平均約6週)8)。14例中有2例發生失明。
GCA與霍納氏症候群
Section titled “GCA與霍納氏症候群”Sverdlichenko等人(2022)報告在53例GCA患者中有2例合併霍納氏症候群(部分眼瞼下垂和瞳孔縮小)5)。推測機制為椎動脈及其分支的血管炎導致腦幹內第一級交感神經元缺血。對於50歲以上新發霍納氏症候群的患者,建議檢查GCA症狀和發炎標記物。
GCA的罕見缺血性併發症
Section titled “GCA的罕見缺血性併發症”GCA可引起視覺以外的多種缺血性併發症3)。
- 腦血管事件:發生率2-7%。
- 舌壞死和頭皮壞死:罕見但嚴重的併發症。
- 周邊動脈併發症。
- Charles Bonnet症候群:永久性視力喪失後出現的慢性視幻覺。在視力障礙患者中報告率為0.4-30%3)。
家族性GCA報告
Section titled “家族性GCA報告”Hayreh等人(2021)報告了一個印度裔5兄弟中有3人患巨細胞動脈炎的病例,提示體染色體隱性遺傳模式7)。這被認為是非白人中首例家族性GCA報告。
8. 參考文獻
Section titled “8. 參考文獻”- Mandura RA. Giant Cell Arteritis Presenting as Unilateral Arteritic Anterior Ischemic Optic Neuropathy. Cureus. 2021;13(7):e16653. doi:10.7759/cureus.16653. PMID:34462685; PMCID:PMC8387793.
- Szydełko-Paśko U, Przeździecka-Dołyk J, Kręcicka J, Małecki R, Misiuk-Hojło M, Turno-Kręcicka A. Arteritic Anterior Ischemic Optic Neuropathy in the Course of Giant Cell Arteritis After COVID-19. The American journal of case reports. 2022;23:e933471. doi:10.12659/AJCR.933471. PMID:35015754; PMCID:PMC8762612.
- Jalaledin DS, Ross C, Makhzoum JP. Rare Ischemic Complications of Giant Cell Arteritis: Case Series and Literature Review. The American journal of case reports. 2022;23:e937565. doi:10.12659/AJCR.937565. PMID:36240129; PMCID:PMC9578098.
- Piccus R, Hansen MS, Hamann S, Mollan SP. An update on the clinical approach to giant cell arteritis. Clinical medicine (London, England). 2022;22(2):107-111. doi:10.7861/clinmed.2022-0041. PMID:35304369; PMCID:PMC8966809.
- Sverdlichenko I, Lam C, Donaldson L, Margolin E. Horner Syndrome in Giant Cell Arteritis: Case Series and Review of the Literature. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2022;42(3):340-345. doi:10.1097/WNO.0000000000001593. PMID:35427255.
- Stewart C, Asif RH, Dakkak T, Singh H, Javaid MA, Patel N. Diagnostic Dilemmas in Giant Cell Arteritis: Overcoming Anchoring Bias. Case reports in rheumatology. 2025;2025:6632374. doi:10.1155/crrh/6632374. PMID:40726518; PMCID:PMC12303627.
- Hayreh SS. Familial giant cell arteritis. BMJ case reports. 2021;14(7). doi:10.1136/bcr-2021-243304. PMID:34301685; PMCID:PMC8311300.
- Yoshimoto K, Kaneda S, Asada M, Taguchi H, Kawashima H, Yoneima R, Matsuoka H, Tsushima E, et al. Giant Cell Arteritis after COVID-19 Vaccination with Long-Term Follow-Up: A Case Report and Review of the Literature. Medicina (Kaunas, Lithuania). 2023;59(12). doi:10.3390/medicina59122127. PMID:38138230; PMCID:PMC10744572.
- Wakabayashi H, Iwayanagi M, Sakai D, et al. Development of giant cell arteritis after vaccination against SARS-CoV2. Medicine. 2023;102(21):e33814.
- Qadir A, Khakwani AS, Khan MR, et al. Ocular Syphilis Mimicking Giant Cell Arteritis. Cureus. 2022;14(9):e29286.