Die arteriitische anteriore ischämische Optikusneuropathie (Arteritic Anterior Ischemic Optic Neuropathy; AAION) wird durch eine Ischämie des Sehnervs aufgrund einer Vaskulitis der versorgenden Gefäße verursacht. Bei der Vaskulitis kommt es durch Verdickung der Gefäßwand, Verengung des Lumens und Thrombose zu einer ischämischen Nekrose. Die Vaskulitis der kurzen hinteren Ziliararterien (SPCAs), die den Sehnervenkopf versorgen, wird als Ursache angesehen. Die AAION macht 5–10 % aller anterioren ischämischen Optikusneuropathien aus, die Mehrheit sind nicht-arteriitische anteriore ischämische Optikusneuropathien (NAION).
Als Grunderkrankung ist die Riesenzellarteriitis (Giant Cell Arteritis; GCA, früher Arteriitis temporalis) am häufigsten. Weitere Ursachen sind Herpes Zoster, rezidivierende Polychondritis, Takayasu-Arteriitis, rheumatoide Arthritis, Polyarteriitis nodosa, SLE und allergische granulomatöse Angiitis (Churg-Strauss-Syndrom).
Überblick und Geschichte der Riesenzellarteriitis (RZA)
Die RZA ist eine systemische granulomatöse Vaskulitis, die mittelgroße bis große Arterien befällt. Die älteste Beschreibung stammt angeblich von Ali Ibn Isa al-Kahhal aus Bagdad im 10. Jahrhundert. 1890 beschrieb Hutchinson schmerzhafte rote Stränge am Kopf, und 1932 führte Bayard Horton die erste Biopsie der Arteria temporalis durch und beschrieb eine granulomatöse Vaskulitis. 1941 beschrieb Gilmour erstmals Riesenzellen, was zum heutigen Krankheitsnamen führte.
Sie tritt häufiger bei Frauen über 50 Jahren auf (Verhältnis Männer:Frauen 1:3), mit einem steilen Anstieg der Inzidenz nach dem 70. Lebensjahr. Das mediane Erkrankungsalter der GCA liegt bei 75 Jahren. Die geschätzte jährliche Inzidenz der AAION beträgt bei über 50-Jährigen 0,36 pro 100.000.
Visuelle Komplikationen der GCA treten bei 10–30 % (in einigen Berichten bis zu 70 %) auf, und die AAION macht 60–90 % der mit GCA verbundenen Sehverluste aus3). Die Inzidenz der GCA steigt mit dem Alter, von 2,3 Fällen pro 100.000 in den 60ern auf 44,7 Fälle pro 100.000 in den 90ern.
Sie ist am häufigsten bei nordischen Kaukasiern (in Norwegen etwa 30 pro 100.000) und selten bei Schwarzen und Asiaten. In Japan beträgt die Inzidenz 1,47 pro 100.000, was im Vergleich zu westlichen Ländern extrem selten ist. In Europa ist sie die häufigste primäre systemische Vaskulitis bei über 50-Jährigen, mit einer jährlichen Inzidenz von 32–290 Fällen pro Million Einwohner6).
QWas ist der Unterschied zwischen AAION und nicht-arteriitischer anteriorer ischämischer Optikusneuropathie (NAION)?
A
Die AAION macht 5–10 % aller anterioren ischämischen Optikusneuropathien aus und wird durch Vaskulitiden wie die Riesenzellarteriitis verursacht. Die Sehprognose ist deutlich schlechter als bei der NAION; über 60 % haben eine Sehschärfe unter 20/200. Bei der NAION zeigt sich am kontralateralen Auge eine „Disc at risk“ (kleine Papille, kleine Exkavation), während bei der AAION der kontralaterale Papillendurchmesser und die physiologische Exkavation normal sind. Für die Differenzialdiagnose sind Entzündungsmarker wie BSG und CRP hilfreich; bei der NAION sind diese nicht erhöht.
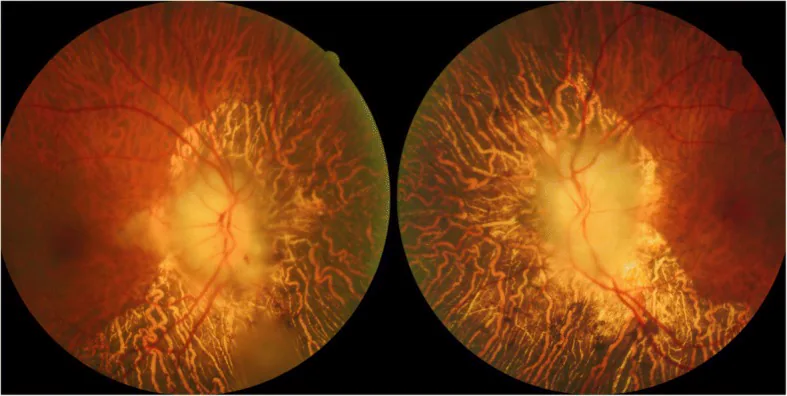
Tian G, et al. Giant cell arteritis presenting as bilateral anterior ischemic optic neuropathy: a biopsy-proven case report in Chinese patient. BMC Ophthalmol. 2018. Figure 1. PMCID: PMC6208180. License: CC BY.
Fundusfotografie bei Erstvorstellung mit schwerem beidseitigem Papillenödem von kreideweißem Aussehen, linearen Blutungen und weichen Exsudaten am rechten Auge sowie diffuser peripapillärer Aderhautatrophie. Dies entspricht dem im Abschnitt „2. Hauptsymptome und klinische Befunde“ beschriebenen „kreideweißen Ödem“.
Bei der GCA manifestiert sich die Erkrankung bei älteren Menschen als plötzlicher Sehverlust auf einem oder beiden Augen. Häufig wird eine Amaurosis fugax als Prodromalsymptom berichtet. Ohne Behandlung tritt innerhalb kurzer Zeit häufig auch eine Beteiligung des anderen Auges auf.
Plötzlicher Sehverlust: Über 60 % erleiden eine schwere Beeinträchtigung mit einer Sehschärfe unter 20/200. Bei über 20 % kommt es zu keiner Lichtwahrnehmung, einer schweren funktionellen Beeinträchtigung.
Amaurosis fugax: Tritt bei etwa 30 % der Fälle mit permanentem Sehverlust als Prodromalsymptom auf, im Durchschnitt 8,5 Tage zuvor. Die berichtete Inzidenz liegt bei 2–19 %. Bei der NAION ist sie äußerst selten, was ein wichtiges Unterscheidungsmerkmal darstellt.
Kopfschmerzen: Das häufigste Allgemeinsymptom, das bei 65–90 % auftritt. Neu aufgetretene temporale oder okzipitale Kopfschmerzen sind charakteristisch1).
Kieferclaudicatio (jaw claudication): Schmerzen oder Ermüdung des Kiefers beim Kauen. Das spezifischste Symptom der GCA. Die Häufigkeit beträgt 11–45 %1).
Kopfhautdruckschmerz: Schmerzhaftigkeit der Temporalarterie und der Kopfhautregion. Unbehagen beim Kämmen der Haare oder beim Auflegen des Kopfes auf das Kissen.
PMR-Symptome (Polymyalgia rheumatica): treten bei bis zu 50 % der Patienten auf. Bilaterale Schmerzen und Steifheit von Nacken, Schultern und Becken.
Doppelbilder (Diplopie): aufgrund von Lähmungen der Hirnnerven III, IV und VI. Tritt bei 10–15 % auf3).
Einschränkung der Augenbeweglichkeit: kann durch Hirnnervenlähmungen verursacht werden.
Okkulte GCA: bis zu 20 % der AAION-Patienten weisen keine offensichtlichen Allgemeinsymptome auf.
QIst eine AAION auch ohne Allgemeinsymptome möglich?
A
Ja. Eine als okkulte Riesenzellarteriitis (okkulte GCA) bezeichnete Erkrankung liegt bei bis zu 20 % der AAION-Patienten vor, ohne typische Allgemeinsymptome wie Kopfschmerzen oder Kieferclaudicatio. Das Fehlen von Allgemeinsymptomen schließt eine Riesenzellarteriitis nicht aus; Blutuntersuchungen (BSG, CRP) und eine Biopsie der Temporalarterie sind erforderlich.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Blasses Papillenödem (pallid swelling): typischer Befund der AAION. Erscheint als kreidig-weiße Blässe (chalky-white pallor), im Gegensatz zum kongestiven Ödem der NAION.
Flammenförmige Blutungen: können peripapillär auftreten.
Cotton-Wool-Flecken: können am hinteren Pol auftreten.
Verengung der peripapillären Netzhautarteriolen.
Zilioretinaler Arterienverschluss: relativ spezifischer Befund der AAION.
Zentraler Netzhautarterienverschluss (ZNAV): kann ebenfalls auftreten.
RAPD positiv: Bei einseitiger oder asymmetrischer Optikusneuropathie ist ein relativer afferenter Pupillendefekt (RAPD) positiv.
Gesichtsfeldausfall: Horizontaler Halbseitenausfall (altitudinal field defect) ist am häufigsten.
Papille des anderen Auges: Normal (Unterschied zum Disc at risk bei NAION).
Optikusatrophie und Papillenexkavation: Innerhalb von 6–8 Wochen fortschreitende Optikusatrophie mit Papillenexkavation. Bei über 90 % der AAION nachweisbar3).
Anomalien der Temporalarterie: Schwellung, Knoten, Druckschmerz, abgeschwächter oder fehlender Puls.
Fluoreszenzangiographie: Verzögerte Füllung der Papille, verzögerte oder fehlende Füllung der peripapillären Aderhaut (segmentale Ischämie) sind charakteristisch.
Bei der Vaskulitis führen Gefäßwandverdickung und Thrombusbildung zu ischämischer Nekrose. Die Vaskulitis der kurzen hinteren Ziliararterien (SPCAs) verursacht eine Ischämie des vorderen Sehnervs, begleitet von segmentaler Ischämie der Aderhaut.
Dendritische Zellen der Gefäßwand fungieren als Hauptbeitragsfaktor der Erkrankung. Über die Vasa vasorum der Adventitia dringen Makrophagen und T-Zellen ein, initiieren eine pathogene Kaskade und verursachen eine granulomatöse Vaskulitis, die mittelgroße bis große Arterien befällt.
Alter : Größter Risikofaktor. Medianes Alter 75 Jahre. Auftreten vor dem 50. Lebensjahr ist äußerst selten.
Geschlecht : 2- bis 6-fach erhöhtes Risiko bei Frauen.
Ethnie : Am häufigsten bei nordischen Kaukasiern. Selten bei Schwarzen und Asiaten.
Genetische Faktoren : HLA-DRB1*04, DRW6, DR3 sind mit erhöhter Suszeptibilität assoziiert. Polymorphismen des TNF-α-Gens und des IL-10-Promotors korrelieren ebenfalls mit erhöhtem Risiko. Es gibt Berichte über familiäre RZA bei Nicht-Kaukasiern 7).
Umwelt- und Infektionsfaktoren : Varizella-Zoster-Virus (VZV), Chlamydia pneumoniae und Parvovirus B19 werden vermutet.
Altersbedingte Mechanismen : Verkalkung der Lamina elastica interna, Elastin und extrazellulärer Matrix könnte die altersspezifische Expression erklären.
Rauchen, niedriger BMI, frühe Menopause : Alle als Risikofaktoren berichtet.
PMR-Koinzidenz : RZA und PMR zeigen eine starke Assoziation.
COVID-19 : Während der Pandemie wurde ein 70%iger Anstieg der RZA-Inzidenz berichtet 2). SARS-CoV-2 hat eine Affinität zum Gefäßendothel, und die Ähnlichkeit mit Vaskulitis legt einen pathogenetischen Zusammenhang nahe.
Die Diagnose der AAION wird parallel zur gesicherten Diagnose der Riesenzellarteriitis durchgeführt. Die Unterscheidung zwischen AAION und NAION ist direkt mit der Behandlungsstrategie verbunden, daher ist eine schnelle und systematische Bewertung wichtig.
BSG : Sensitivität 86 %. Kann 70–120 mm/h erreichen. Normalwerte: Männer = Alter/2, Frauen = (Alter+10)/2. Allerdings zeigen bis zu 10 % normale Werte.
CRP : Sensitivität 97,5 %. Spezifischer als BSG. In den ACR/EULAR-Klassifikationskriterien von 2022 wurde CRP ≥ 10 mg/L als zusätzliches Item aufgenommen 4).
BSG+CRP kombiniert : Sensitivität 99,2 %, Spezifität 97 %. Akute-Phase-Proteine sind bei über 80 % erhöht.
Sonstiges : Die Kombination aus Thrombozytose, CRP und Thrombozyten hat den höchsten diagnostischen Nutzen (p < 0,001) 4).
Temporalarterienbiopsie (TAB)
Stellenwert : Goldstandard für die definitive Diagnose einer Riesenzellarteriitis. Bei korrekter Durchführung Sensitivität und Spezifität > 95 %.
Positive Befunde : Intimaverdickung, Ruptur der Lamina elastica interna, chronisches entzündliches Infiltrat mit Riesenzellen. Für die pathologische Bestätigung sind Zerstörung der Lamina elastica interna und Entzündungszellinfiltrat (akute Phase) oder Fibrose (chronische Phase) erforderlich. Riesenzellen sind für die definitive Diagnose nicht zwingend erforderlich.
Falsch-negative : Falsch-negative Rate von 3–5 % aufgrund von Skip-Läsionen. Es gibt Berichte von bis zu 61 % 6). Ein negatives TAB schließt eine GCA nicht aus.
Zeitpunkt der Durchführung : Die Biopsie sollte innerhalb weniger Tage nach Beginn der Steroidtherapie erfolgen.
Bildgebende Diagnostik
Temporalarterien-Ultraschall (CDUS) : Nicht-invasiv, wiederholbar. Sensitivität 77 %, Spezifität 96 % 4). Halo-Zeichen (hypoechogener Ring durch Gefäßwandverdickung), Kompressionszeichen, Stenose und Verschluss sind charakteristische Befunde. Aufgrund von Skip-Läsionen ist eine beidseitige und mehrregionale umfassende Untersuchung wichtig 4). Bei beidseitig positivem Halo-Zeichen steigt die Spezifität auf 100 % 4).
PET-CT : Kann bei Großgefäß-GCA (LV-GCA) abnorme Anreicherungen in der Aorta und ihren Ästen nachweisen. Die GAPS-Studie ergab eine Sensitivität von 92 % und eine Spezifität von 85 % 6).
MRT : Hilfreich zur Unterscheidung von AAION und NAION. Kontrastmittelanreicherung der Sehnervenscheide und des orbitalen Fetts (central bright spot) prüfen.
Augenärztliche Untersuchungen
Fluoreszenzangiographie (FA) : Verzögerte Füllung der Papille, verzögerte/defekte Füllung der peripapillären Aderhaut (segmentale Ischämie) sind charakteristisch für AAION. Kann vor Auftreten eines Papillenödems beobachtet werden. Wichtiges Unterscheidungsmerkmal zur NAION.
OCT/OCTA : Nützlich zur Beurteilung eines segmentalen Papillenödems, der Dicke der retinalen Nervenfaserschicht (RNFL) und des ischämischen Zustands der Papille.
ACR-Klassifikationskriterien (1990) und ACR/EULAR-Klassifikationskriterien 2022
Abgrenzung zu anderen Vaskulitiden wie Polyarteriitis nodosa, Granulomatose mit Polyangiitis (Wegener) und SLE. Ein wichtiger Unterscheidungspunkt ist, dass die GCA Lunge und Nieren nicht befällt. Okuläre Syphilis kann GCA-ähnliche Symptome hervorrufen 10). Auch auf okkulte GCA (ca. 20 %) ohne systemische Symptome ist zu achten.
QKann eine negative Biopsie der Temporalarterie eine Riesenzellarteriitis ausschließen?
A
Nein. Aufgrund von Skip-Läsionen (Entzündung nur in einem Teil des Gefäßes) beträgt die Falsch-negativ-Rate 3–5 % (einige Berichte geben bis zu 61 % an 6)). Selbst bei negativer TAB sollte bei klinischem Verdacht auf GCA mit erhöhter BSG und CRP die Behandlung fortgesetzt werden. Die Gesamtbeurteilung von klinischen Befunden, Blutuntersuchungen und Ultraschallbefunden ist wichtig.
Bei Verdacht auf Sehstörung sollte die Behandlung sofort eingeleitet werden, ohne auf die Bestätigung durch Biopsie zu warten. Hauptziel der Behandlung ist die Prävention des Befalls des anderen Auges; eine Verbesserung der Sehkraft des betroffenen Auges ist kaum zu erwarten. Nur bei 15–20 % der Patienten bessert sich das Sehvermögen. Eine stationäre hochdosierte Steroid-Infusionstherapie ist wünschenswert.
Akutphase: Sofortige intravenöse Gabe von Methylprednisolon 1 g/Tag für 3–5 Tage.
Erhaltungsphase: Umstellung auf orales Prednisolon 1 mg/kg/Tag.
Ausschleichen: Langsame Reduktion über mindestens 4–6 Monate, abhängig vom Allgemeinzustand und der BSG. In manchen Fällen kann mehr als ein Jahr erforderlich sein.
Achtung: Eine alternative tägliche Steroidgabe wird nicht empfohlen.
Bei Fehlen von Augen- oder ZNS-Symptomen kann mit Prednisolon 30–40 mg/Tag begonnen werden.
In Betracht ziehen, wenn Nebenwirkungen der langfristigen Steroidanwendung (Cushing-ähnliches Syndrom, Hyperglykämie, Osteoporose, gastrointestinale Symptome usw., die bei etwa 60 % auftreten) problematisch werden.
Tocilizumab (Tocilizumab; IL-6-Rezeptor-Inhibitor): 2017 von der FDA zur Behandlung der Riesenzellarteriitis (GCA) zugelassen. Randomisierte kontrollierte Studien (RCTs) haben einen steroidsparenden Effekt und eine Wirksamkeit bei der Erreichung einer Remission über 12 Monate gezeigt4). Es gibt Berichte über Wirksamkeit bei steroidresistenter AAION. Auch bei GCA-Fällen nach COVID-19-Impfung wurde die Anwendung von TCZ 162 mg subkutan berichtet8)9).
Methotrexat: Erhöht die Rate des dauerhaften Absetzens von Steroiden und verringert das Rückfallrisiko. In Fallberichten wird eine Dosis von 15 mg/Woche in Kombination verwendet1). Bei Rückfall kann eine Steroid-Dosiserhöhung plus MTX-Kombination empfohlen werden.
Niedrig dosiertes Aspirin: Kann zur Prävention kardialer und zerebrovaskulärer ischämischer Komplikationen in Betracht gezogen werden.
QKann die Sehkraft durch eine Steroidbehandlung wiederhergestellt werden?
A
Eine Verbesserung der Sehkraft des betroffenen Auges ist kaum zu erwarten. Nur bei 15–20 % bessert sich die Sehkraft, in den meisten Fällen bleibt eine Sehminderung bestehen. Hauptziel der Steroidbehandlung ist die Verhinderung einer Beteiligung des anderen Auges.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Die Pathophysiologie der Riesenzellarteriitis beruht auf zwei Hauptmechanismen der Immunantwort. Es handelt sich um eine T-Zell-vermittelte granulomatöse Vaskulitis, die durch die Aktivierung dendritischer Zellen in der Gefäßwand ausgelöst wird und selektiv mittelgroße bis große Arterien befällt.
Immun- und Entzündungskaskade (Zwei-Achsen-Modell)
Zerstörung der Gefäßwand : Zerstörung der Lamina elastica interna durch Metalloproteinasen und reaktive Sauerstoffspezies.
Nichtbeteiligung von B-Zellen : Eine Beteiligung von B-Zellen wurde nicht bestätigt, was ein wichtiger Unterscheidungspunkt zur ANCA-assoziierten Vaskulitis ist.
Die Blutversorgung des Sehnervs erfolgt hauptsächlich durch die kurzen hinteren Ziliararterien (SPCAs) und Äste der zentralen Netzhautarterie.
Die SPCAs versorgen die prälaminäre und laminäre Region und tragen auch zur peripapillären Aderhautzirkulation bei.
Bei der GCA führt der thrombotische Verschluss der SPCAs (in 20% der Fälle besonders betroffen) zu einer Ischämie der Sehnervenpapille.
Postmortem-Untersuchungen der akuten AAION haben ein Papillenödem mit Nekrose der prälaminären, laminären und retrolaminären Region sowie chronische Entzündungszellinfiltration bestätigt.
Fluoreszenzangiographiedaten stützen die histopathologischen Belege für eine Beteiligung der SPCAs.
Charakteristisch ist ein Riss der Lamina elastica interna, und die Riesenzellen befinden sich in der Nähe der gerissenen Lamina elastica interna. In der akuten Phase überwiegt die lymphozytäre Infiltration, in der chronischen Phase kommt es zur Fibrose. Als Heilungsreaktion auf die Entzündung treten Intimaverdickung, Myofibroblastenproliferation und Ablagerung extrazellulärer Matrix auf, was zu Gefäßstenose und -verschluss führt.
7. Aktuelle Forschung und zukünftige Perspektiven (Berichte aus der Forschungsphase)
Der orale selektive JAK1-Inhibitor Upadacitinib wurde 2025 von der FDA zur Behandlung der Riesenzellarteriitis (RZA) zugelassen. Er wird als neue Behandlungsoption angesehen, die auf den IL-6–JAK–STAT-Signalweg abzielt.
Tocilizumab hat in randomisierten kontrollierten Studien (RCTs) einen GC-sparenden Effekt und eine Wirksamkeit bei der Erreichung einer Remission über 12 Monate gezeigt 4) und etabliert sich als alternative Therapie zur Reduzierung der Toxizität einer langfristigen GC-Gabe.
Der ultraschallgestützte Schnelldiagnosepfad für Riesenzellarteriitis verbreitet sich in Europa. Die Einführung des Fast-Track-Pfads hat eine Verringerung des Sehverlusts, eine Eindämmung der Überbehandlung und eine Verbesserung der Kosteneffektivität gezeigt 4). Ultraschall ist nicht-invasiv, wiederholbar und ermöglicht die gleichzeitige Beurteilung mehrerer Arterienregionen, was ihn zu einem wichtigen Instrument für die Frühdiagnose macht.
Ein Subtyp der Riesenzellarteriitis, der vor dem Hintergrund eines myelodysplastischen Syndroms (MDS) auftritt (GCA-MDS), wird anerkannt. Bei 10–20 % der MDS-Patienten entwickelt sich eine Autoimmunerkrankung. GCA-MDS könnte eine geringere Prävalenz klassischer Symptome (Kopfschmerzen, Kieferclaudicatio, AAION usw.) aufweisen als die typische RZA und neigt zu einer Steroidabhängigkeit mit verringerter steroidfreier und rezidivfreier Überlebensrate. Die Zugabe von hypomethylierenden Wirkstoffen (Azacitidin/Decitabin) könnte vorteilhaft sein; eine prospektive Studie (NCT02985190) läuft. Der größte Bericht ist eine französische multizentrische Serie mit 21 Fällen aus dem Jahr 2019.
Es gibt Berichte über einen Anstieg der RZA-Inzidenz um 70 % während der COVID-19-Pandemie 2). Mehrere Berichte zeigen einen Anstieg der RZA-Fälle im Jahr 2020, eine erhöhte Rate an Augenkomplikationen und vermuten eine Beteiligung von Endothelschäden, Th1-Immunität und des Monozyten-Makrophagen-Systems 2). Es gibt auch Fallberichte, die darauf hindeuten, dass SARS-CoV-2 die RZA ausgelöst haben könnte.
Mehrere Fälle von GCA nach COVID-19-Impfung wurden berichtet. Yoshimoto et al. (2023) überprüften 14 Fälle und berichteten, dass der Zeitraum vom Beginn bis zur Diagnose 2 Wochen bis 4 Monate (durchschnittlich etwa 6 Wochen) betrug 8). In 2 der 14 Fälle trat Erblindung auf.
Sverdlichenko et al. (2022) berichteten über eine Koinzidenz des Horner-Syndroms (partielle Ptosis und Miosis) bei 2 von 53 GCA-Patienten 5). Der vermutete Mechanismus ist eine Ischämie der ersten sympathischen Neuronen im Hirnstamm aufgrund einer Vaskulitis der Arteria vertebralis und ihrer Äste. Bei neu aufgetretenem Horner-Syndrom bei über 50-Jährigen wird empfohlen, auf GCA-Symptome zu achten und Entzündungsmarker zu testen.
Hayreh et al. (2021) berichteten über einen Fall, bei dem 3 von 5 Brüdern indischer Abstammung eine Riesenzellarteriitis entwickelten, was auf ein autosomal-rezessives Vererbungsmuster hindeutet 7). Dies wird als erster Bericht über familiäre GCA bei Nicht-Weißen angesehen.
Szydełko-Paśko U, Przeździecka-Dołyk J, Kręcicka J, Małecki R, Misiuk-Hojło M, Turno-Kręcicka A. Arteritic Anterior Ischemic Optic Neuropathy in the Course of Giant Cell Arteritis After COVID-19. The American journal of case reports. 2022;23:e933471. doi:10.12659/AJCR.933471. PMID:35015754; PMCID:PMC8762612.
Jalaledin DS, Ross C, Makhzoum JP. Rare Ischemic Complications of Giant Cell Arteritis: Case Series and Literature Review. The American journal of case reports. 2022;23:e937565. doi:10.12659/AJCR.937565. PMID:36240129; PMCID:PMC9578098.
Piccus R, Hansen MS, Hamann S, Mollan SP. An update on the clinical approach to giant cell arteritis. Clinical medicine (London, England). 2022;22(2):107-111. doi:10.7861/clinmed.2022-0041. PMID:35304369; PMCID:PMC8966809.
Sverdlichenko I, Lam C, Donaldson L, Margolin E. Horner Syndrome in Giant Cell Arteritis: Case Series and Review of the Literature. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2022;42(3):340-345. doi:10.1097/WNO.0000000000001593. PMID:35427255.
Stewart C, Asif RH, Dakkak T, Singh H, Javaid MA, Patel N. Diagnostic Dilemmas in Giant Cell Arteritis: Overcoming Anchoring Bias. Case reports in rheumatology. 2025;2025:6632374. doi:10.1155/crrh/6632374. PMID:40726518; PMCID:PMC12303627.
Yoshimoto K, Kaneda S, Asada M, Taguchi H, Kawashima H, Yoneima R, Matsuoka H, Tsushima E, et al. Giant Cell Arteritis after COVID-19 Vaccination with Long-Term Follow-Up: A Case Report and Review of the Literature. Medicina (Kaunas, Lithuania). 2023;59(12). doi:10.3390/medicina59122127. PMID:38138230; PMCID:PMC10744572.
Wakabayashi H, Iwayanagi M, Sakai D, et al. Development of giant cell arteritis after vaccination against SARS-CoV2. Medicine. 2023;102(21):e33814.
Qadir A, Khakwani AS, Khan MR, et al. Ocular Syphilis Mimicking Giant Cell Arteritis. Cureus. 2022;14(9):e29286.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.