Das Horner-Syndrom ist ein Syndrom, das aufgrund einer Störung des okulären sympathischen Nervensystems vielfältige Augen- und Allgemeinsymptome zeigt. Die Trias besteht aus Miosis, Ptosis und Anhidrose, und die Störung kann zentral, präganglionär oder postganglionär auftreten.
Der Name der Erkrankung leitet sich von Claude Bernard (1852) und Johann Friedrich Horner (1869) ab. Sie wird auch als Bernard-Horner-Syndrom bezeichnet. Der Verlauf des okulären Sympathikus ist lang, vom Hypothalamus über drei Neuronen zum Pupillendilatatormuskel, und je nach Ort der Läsion unterscheiden sich Ursache und Dringlichkeit erheblich.
Die Miosis ist mäßig, und die Anisokorie ist im Dunkeln am deutlichsten. Die Pupillenlichtreaktion bleibt normal, was ein wichtiger Unterschied zur Adie-Pupille oder zur Okulomotoriusparese ist. Die Denervierung des Müllerschen Muskels des Oberlids führt zu einer leichten Ptosis von etwa 2 mm, und die Schädigung des Müllerschen Muskels des Unterlids führt zu einer leichten Anhebung des Unterlids (Upside-down-Ptosis). Zusammen ergeben sie eine verengte Lidspalte und eine scheinbare Enophthalmie.
QIst die Ptosis beim Horner-Syndrom schwerwiegend?
A
In der Regel handelt es sich um eine leichte Ptosis von etwa 2 mm, die durch eine sympathische Denervierung des Müllerschen Muskels des Oberlids verursacht wird. Auch das Unterlid ist leicht angehoben (Upside-down-Ptosis), was zu einer verengten Lidspalte und einer scheinbaren Enophthalmie führt. Die Abgrenzung zu einer kompletten Ptosis (Okulomotoriusparese) ist wichtig.
Li XM, et al. Neuro-ophthalmic observation and 16-month follow-up of horner syndrome after thyroidectomy: A case report. Medicine (Baltimore). 2026. Figure 2. PMCID: PMC12826322. License: CC BY.
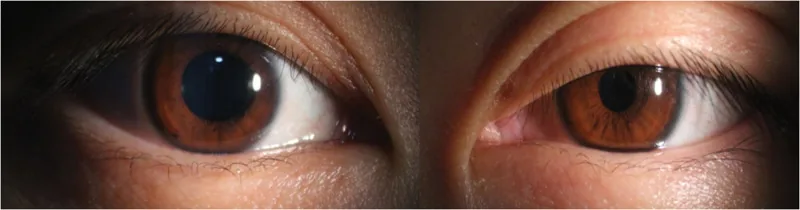
Äußeres Foto beider Augen, das eine leichte Ptosis und Miosis eines Auges zeigt. Veranschaulicht visuell die typischen klinischen Befunde des Horner-Syndroms und eignet sich zur Beschreibung der Hauptsymptome und klinischen Befunde.
Miosis : mäßige Miosis. Im Dunkeln ist die Anisokorie zum gesunden Auge am deutlichsten. Der Lichtreflex bleibt normal.
Ptosis : leichte Ptosis von etwa 2 mm durch Störung des Müller-Muskels des Oberlids. Störung des Müller-Muskels des Unterlids führt zu Anhebung des Unterlids (Upside-down-Ptosis) → Verengung der Lidspalte.
Hypohidrose : Das Ausmaß variiert je nach Läsionsort und ist direkt für die topische Diagnose relevant. Kann mit Hitzegefühl und Rötung im Bereich der Hypohidrose einhergehen.
Bei angeborenem Auftreten oder innerhalb des ersten Lebensjahres liegt eine Iris-Hypoplasie (Heterochromie) vor. Ursachen können Geburtstraumata der Halswirbelsäule (Zangengeburt) sein, viele Fälle bleiben jedoch idiopathisch.
Raeder-Syndrom: Trigeminusneuralgie + postganglionäres Horner-Syndrom. Hinweis auf eine Läsion in der Nähe des Ganglion trigeminale; Abklärung auf Aneurysma der A. carotis interna, Tumor der mittleren Schädelgrube oder Nasopharynxtumor erforderlich.
Alternierendes Horner-Zeichen: Die betroffene Seite wechselt in einem nahezu regelmäßigen Zyklus (alle 1–3 Tage). Tritt während des Nachtschlafs auf. Assoziationen mit Shy-Drager-Syndrom, Halswirbelsäulentrauma und Multipler Sklerose wurden berichtet.
Typische Ursachen : Dissektion der A. carotis interna, nach Schilddrüsenoperation, nach Karotisendarteriektomie, traumatische Dissektion der A. carotis interna
Merkmale : Akuter Beginn mit Nackenschmerz oder Kopfschmerz ist ein starker Hinweis auf eine Dissektion, ein Notfall.
Die GCA ist eine granulomatöse Vaskulitis mittlerer und großer Gefäße, die nach dem 50. Lebensjahr auftritt, und es wurde berichtet, dass sie mit einem Horner-Syndrom einhergehen kann 1). In einer Analyse einer Fallserie von GCA-Patienten wurde bei 2 von 53 Patienten (ca. 4 %) ein Horner-Syndrom festgestellt 1).
In einem Fallbericht hatte ein 67-jähriger Mann eine rechtseitige Trochlearisparese und ein linksseitiges Horner-Syndrom mit einer BSG von 70 mm/h und einem CRP von 10 mg/L. Nach Beginn der Behandlung mit Prednison 60 mg verschwand die Diplopie, aber das Horner-Syndrom blieb bestehen 1). Ein weiterer 71-jähriger Mann hatte bilaterale Kopfschmerzen, Kieferschmerzen, ein linksseitiges Horner-Syndrom und eine Skew Deviation mit einer BSG von 68 mm/h und einem CRP von 46 mg/L. Drei Tage nach Beginn der Behandlung mit Prednison 60 mg verschwanden alle Symptome einschließlich des Horner-Syndroms 1). In beiden Fällen bestätigte die Biopsie der Temporalarterie die GCA, und MRT/MRA zeigten keine Auffälligkeiten 1).
1% Apraclonidinhydrochlorid (Iopidine®) in beide Augen tropfen (Off-Label-Anwendung).
Normales Auge: Keine Reaktion oder leichte Miosis aufgrund der vorherrschenden α2-Rezeptorwirkung
Horner-Auge: Denervierungsüberempfindlichkeit führt zu einer Zunahme der α1-Rezeptoren → Mydriasis 30–60 Minuten nach der Tropfengabe → Umkehrung der Anisokorie
Sensitivität: 88–100 %
Spezifität: 100 %
Hinweis: Wenn seit dem Auftreten weniger als 3 Tage vergangen sind, hat sich die Denervierungsüberempfindlichkeit noch nicht entwickelt, was zu einem falsch-negativen Ergebnis führen kann.
Der 5% Kokain-Test nutzt die Hemmung der Noradrenalin-Wiederaufnahme, um das Vorhandensein einer Mydriasis zu beurteilen. Bei einem normalen Auge tritt nach 90–120 Minuten eine Mydriasis (++) auf, während bei einem Horner-Auge die Mydriasis unzureichend ist.
Differenzialdiagnose der Läsionsstelle (pharmakologische Lokalisationsdiagnostik)
Tyramin bewirkt die Freisetzung von Noradrenalin aus den Nervenendigungen. Bei präganglionären Läsionen verbleibt Noradrenalin in den Nervenendigungen, was zu einer Pupillenerweiterung führt. Bei postganglionären Läsionen verbleibt es nicht, daher keine Pupillenerweiterung.
Auch die Gabe von Hydroxyamphetamin (1%) Augentropfen ermöglicht die Unterscheidung zwischen prä- und postganglionären Läsionen: Bei präganglionären Läsionen kommt es zu einer Pupillenerweiterung (+), bei postganglionären zu keiner (−)2).
QKann der Apraclonidin-Test jederzeit durchgeführt werden?
A
Da die Denervierungsüberempfindlichkeit erst nach mindestens 3 Tagen nach Beginn ausreichend ausgeprägt ist, kann der Test in der akuten Phase (innerhalb von 3 Tagen nach Beginn) falsch negativ ausfallen. In der akuten Phase wird der Kokain-Augentropfen-Test empfohlen, aber in Japan kann es schwierig sein, Kokain-Augentropfen zu erhalten.
Führen Sie bildgebende Untersuchungen entsprechend der Läsionsstelle durch. Für jede Läsionsstelle sind entsprechende bildgebende Verfahren erforderlich, aber die Thoraxbildgebung hat Priorität, um Lungenkrebs und Mediastinaltumoren auszuschließen.
Postganglionär : MRT/MRA des Halses (dringender Ausschluss einer Karotisdissektion)
Zentral : MRT des Kopfes (Suche nach Hirnstammläsionen)
Kind : CT/MRT von Abdomen und Thorax + Urin-Katecholamine (Ausschluss eines Neuroblastoms)
Die Behandlung der Grunderkrankung hat höchste Priorität. Liegen keine anderen systemischen Befunde vor und wird die Erkrankung als gutartig eingestuft, erfolgt eine Beobachtung.
Privina® Augentropfen (Naphazolin) : Bei peripherer Ptosis (mit Denervierungsüberempfindlichkeit) kann sich der Müller-Muskel zusammenziehen und die Ptosis verbessern (Off-Label-Use).
Reparatur der Levator-Aponeurose : Wirksam bei Ptosis von etwa 2 mm.
Karotisdissektion (akutes Horner + Nackenschmerzen/Kopfschmerzen) : Sofortige Überweisung an Neurologie/Neurochirurgie. Antithrombotische Therapie (Antikoagulation oder Thrombozytenaggregationshemmung) erwägen.
Pancoast-Tumor (Spitzenverschattung der Lunge) : Überweisung an Onkologie.
Kind / Neuroblastom (erworbenes Horner bei Kindern) : Überweisung an Pädiatrie/Kinderchirurgie.
GCA (Entzündungsmarker erhöht bei über 50-Jährigen): sofort hochdosiertes orales Prednison (60 mg/Tag) beginnen 1), um einen irreversiblen Sehverlust zu verhindern.
QBessert sich die Miosis nach der Behandlung des Horner-Syndroms?
A
Wenn die Grunderkrankung behandelt wird, variieren die Erholungschancen je nach Ort und Ursache der Läsion. Iatrogene (postoperative) und idiopathische Fälle haben oft einen gutartigen Verlauf. Eine Karotisdissektion kann sich durch Notfallbehandlung bessern. Tumoröse Ursachen hängen von der Prognose des Tumors selbst ab. Bei GCA-Fällen gibt es Berichte über eine Besserung der Symptome durch Prednison, aber in einigen Fällen kann das Horner-Syndrom bestehen bleiben.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
1. Neuron (zentral): Hypothalamus → Abstieg im Hirnstamm → Synapse im Budge-Zentrum (Ziliospinalzentrum, C8-T2 Seitenhorn)
2. Neuron (präganglionär): Budge-Zentrum → verlässt das Rückenmark, passiert die Lungenspitze → Synapse im Ganglion stellatum (Ganglion cervicale superius)
3. Neuron (postganglionär): Ganglion stellatum → aufsteigend entlang der A. carotis interna → durch den Sinus cavernosus → als Nn. ciliares longi zum M. dilatator pupillae und M. Müller
Da dieser Weg lang ist, können verschiedene Ursachen vom Hirnstamm bis zur Lungenspitze und zum Hals beteiligt sein.
Sympathische Innervation des M. Müller und Mechanismus der Symptomentstehung
Der M. Müller des Oberlids (verantwortlich für etwa 2 mm Lidhebung) und der M. Müller des Unterlids werden sympathisch innerviert. Denervation führt zu einer Ptosis des Oberlids (ca. 2 mm) und einer Anhebung des Unterlids (Upside-down-Ptosis), was eine Verengung der Lidspalte und eine scheinbare Enophthalmie verursacht.
Bei postganglionären Läsionen wird die Freisetzung von Noradrenalin aus den Nervenendigungen unterbrochen. Dies führt zu einer kompensatorischen Hochregulation (Up-Regulation) der α1-Rezeptoren des M. dilatator pupillae. Selbst eine geringe α1-Wirkung (α1-Wirkung von Apraclonidin) führt zur Pupillenerweiterung, was das Prinzip der pharmakologischen Diagnostik darstellt. Die Entwicklung dieser Überempfindlichkeit dauert mindestens 3 Tage.
Es wird angenommen, dass die Vaskulitis der GCA auf die perforierenden Äste der A. vertebralis übergreift, wodurch die Durchblutung des Hirnstamms reduziert wird und eine Schädigung des ersten Neurons auftritt 1). Alternativ kann die granulomatöse Entzündung der Wand der A. carotis interna direkt den sympathischen Plexus schädigen und eine postganglionäre Läsion verursachen 1). Literaturübersichten berichten sowohl über zentrale als auch postganglionäre Läsionen, wobei der Ort der Läsion von Fall zu Fall variiert 1).
Berichte über Horner-Syndrom in Verbindung mit GCA sind in der Literatur selten (weniger als 10 Fälle), und die Details des Pathomechanismus sind noch nicht geklärt1). Von den 8 zuvor berichteten Fällen besserten sich die systemischen Symptome in allen Fällen unter Prednison 60 mg/Tag (ca. 1 mg/kg/Tag). Die Erholung des Horner-Syndroms variierte von Fall zu Fall; einige Fälle verschwanden vollständig 3 Tage nach Behandlungsbeginn, andere blieben bestehen1). Zentrale Läsionen (erstes Neuron) sind wahrscheinlich auf eine Hirnstammischämie zurückzuführen und gehen häufig mit einer Trochlearisparese oder einer Skew Deviation einher1). In vielen Fällen sind mittels MRT/MRA keine Auffälligkeiten nachweisbar, und die Diagnose stützt sich auf klinische Befunde und Entzündungsmarker1).
Sverdlichenko I, Lam C, Donaldson L, Margolin E. Horner Syndrome in Giant Cell Arteritis: Case Series and Review of the Literature. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2022;42(3):340-345. doi:10.1097/WNO.0000000000001593. PMID:35427255.
Martin TJ. Horner Syndrome: A Clinical Review. ACS chemical neuroscience. 2018;9(2):177-186. doi:10.1021/acschemneuro.7b00405. PMID:29260849.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.