La neuropathie optique ischémique antérieure artéritique (Arteritic Anterior Ischemic Optic Neuropathy; AAION) est due à une ischémie du nerf optique causée par une vascularite des vaisseaux nourriciers. La vascularite entraîne un épaississement de la paroi vasculaire, un rétrécissement de la lumière et une thrombose, conduisant à une nécrose ischémique. La vascularite des artères ciliaires postérieures courtes (SPCAs) qui irriguent la tête du nerf optique en est la cause principale. La NOIAA représente 5 à 10 % de toutes les neuropathies optiques ischémiques antérieures, la majorité étant des neuropathies optiques ischémiques antérieures non artéritiques (NOIAN).
La maladie sous-jacente la plus fréquente est l’artérite à cellules géantes (Giant Cell Arteritis; GCA, anciennement artérite temporale). D’autres causes incluent le zona, la polychondrite récidivante, l’artérite de Takayasu, la polyarthrite rhumatoïde, la périartérite noueuse, le lupus érythémateux disséminé (LED) et la granulomatose éosinophilique avec polyangéite (syndrome de Churg-Strauss).
Aperçu et historique de l’artérite à cellules géantes (ACG)
L’ACG est une vascularite granulomateuse systémique touchant les artères de moyen et gros calibre. La plus ancienne description remonterait à Ali Ibn Isa al-Kahhal à Bagdad au 10e siècle. En 1890, Hutchinson a décrit des cordons rouges douloureux sur la tête, et en 1932, Bayard Horton a réalisé la première biopsie de l’artère temporale, décrivant une vascularite granulomateuse. En 1941, Gilmour a décrit pour la première fois des cellules géantes, ce qui a établi le nom actuel de la maladie.
Elle touche principalement les femmes de plus de 50 ans (ratio hommes/femmes 1:3), avec une augmentation brutale de l’incidence après 70 ans. L’âge médian d’apparition de la GCA est de 75 ans. L’incidence annuelle estimée de l’AAION chez les personnes de plus de 50 ans est de 0,36 pour 100 000 habitants.
Les complications visuelles de la GCA surviennent dans 10 à 30 % des cas (jusqu’à 70 % selon certaines études), et l’AAION représente 60 à 90 % des pertes de vision associées à la GCA3). L’incidence de la GCA augmente avec l’âge, passant de 2,3 cas pour 100 000 personnes dans la soixantaine à 44,7 cas pour 100 000 dans la quatre-vingt-dizaine.
Elle est plus fréquente chez les Blancs d’origine nordique (environ 30 personnes pour 100 000 en Norvège) et rare chez les Noirs et les Asiatiques. Au Japon, l’incidence est de 1,47 pour 100 000, extrêmement rare par rapport à l’Occident. En Europe, c’est la vascularite systémique primitive la plus fréquente chez les personnes de plus de 50 ans, avec une incidence annuelle de 32 à 290 cas par million d’habitants6).
QQuelle est la différence entre l'AAION et la neuropathie optique ischémique antérieure non artéritique (NAION) ?
A
L’AAION représente 5 à 10 % de toutes les neuropathies optiques ischémiques antérieures et est causée par une vascularite telle que l’artérite à cellules géantes. Le pronostic visuel est nettement moins bon que celui de la NAION, avec plus de 60 % des patients ayant une acuité visuelle inférieure à 20/200. Dans la NAION, on observe un « disque à risque » (petite papille, petite excavation) dans l’œil controlatéral, alors que dans l’AAION, le diamètre papillaire et l’excavation physiologique de l’œil controlatéral sont normaux. Les marqueurs inflammatoires tels que la VS et la CRP sont utiles pour le diagnostic différentiel ; ils ne sont pas élevés dans la NAION.
Tian G, et al. Giant cell arteritis presenting as bilateral anterior ischemic optic neuropathy: a biopsy-proven case report in Chinese patient. BMC Ophthalmol. 2018. Figure 1. PMCID: PMC6208180. License: CC BY.
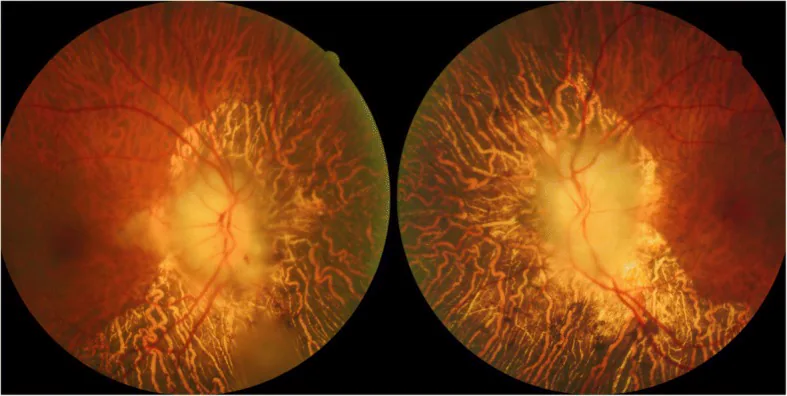
Photographie du fond d’œil initial montrant un œdème papillaire bilatéral sévère avec un aspect blanc crayeux, des hémorragies linéaires et des exsudats mous dans l’œil droit, ainsi qu’une atrophie choroïdienne diffuse péripapillaire. Cela correspond à l’« œdème blanc crayeux » décrit dans la section « 2. Principaux symptômes et signes cliniques ».
Dans la GCA, la maladie se manifeste chez les personnes âgées par une baisse brutale de la vision d’un œil ou des deux yeux. Un amaurose fugace est souvent rapporté comme symptôme prodromique. Sans traitement, l’atteinte de l’œil controlatéral survient fréquemment en peu de temps.
Baisse brutale de la vision : plus de 60 % des patients présentent une déficience sévère avec une acuité visuelle inférieure à 20/200. Plus de 20 % évoluent vers une absence de perception lumineuse, une altération fonctionnelle grave.
Amaurose fugace : survient comme symptôme prodromique dans environ 30 % des cas de perte de vision permanente, en moyenne 8,5 jours avant. L’incidence rapportée est de 2 à 19 %. Elle est extrêmement rare dans la NAION, ce qui constitue un point de différenciation important.
Céphalées : symptôme général le plus fréquent, présent dans 65 à 90 % des cas. Les céphalées temporales ou occipitales d’apparition récente sont caractéristiques1).
Claudication de la mâchoire (jaw claudication) : douleur ou fatigue de la mâchoire lors de la mastication. C’est le symptôme le plus spécifique de la GCA. Sa fréquence est de 11 à 45 %1).
Sensibilité du cuir chevelu : douleur à la palpation de l’artère temporale et de la région du cuir chevelu. Inconfort lors du brossage des cheveux ou en posant la tête sur l’oreiller.
Symptômes généraux : fièvre, perte de poids, fatigue, perte d’appétit, douleurs musculaires, douleurs articulaires, etc.
Symptômes de la PMR (polymyalgie rhumatismale) : surviennent chez jusqu’à 50 % des patients. Douleur et raideur bilatérales du cou, des épaules et du bassin.
Diplopie : due à une paralysie des nerfs crâniens III, IV et VI. Survient dans 10 à 15 % des cas3).
Limitation des mouvements oculaires : peut survenir en raison d’une paralysie des nerfs crâniens.
GCA occulte : jusqu’à 20 % des patients atteints de NAIA ne présentent aucun symptôme systémique évident.
QUne NAIA est-elle possible même en l'absence de symptômes systémiques ?
A
Oui. Une condition appelée artérite à cellules géantes occulte (GCA occulte) est présente chez jusqu’à 20 % des patients atteints de NAIA, sans symptômes systémiques typiques tels que maux de tête ou claudication de la mâchoire. L’absence de symptômes systémiques n’exclut pas une artérite à cellules géantes ; des analyses sanguines (VS, CRP) et une biopsie de l’artère temporale sont essentielles.
Signes cliniques (constatations du médecin lors de l’examen)
Œdème papillaire pâle (pallid swelling) : signe typique de la NAIA. Se présente comme un œdème blanc crayeux (chalky-white pallor), contrastant avec l’œdème congestif de la NAIN.
Hémorragies en flammèche : peuvent être observées autour de la papille.
Taches cotonneuses (cotton wool spots) : peuvent être observées au pôle postérieur.
Rétrécissement des artérioles rétiniennes péripapillaires.
Occlusion de l’artère cilio-rétinienne : signe relativement spécifique de la NAIA.
Occlusion de l’artère centrale de la rétine (OACR) : peut également survenir.
Déficit du champ visuel : l’hémianopsie altitudinale (altitudinal field defect) est la plus fréquente.
Papille de l’œil controlatéral : normale (différence avec le disc at risk de la NAION).
Atrophie optique et excavation papillaire : l’atrophie optique progresse en 6 à 8 semaines avec excavation papillaire. Présente dans plus de 90% des AAION3).
Anomalies de l’artère temporale : distension, nodularité, sensibilité, diminution ou abolition du pouls.
Angiographie à la fluorescéine : retard de remplissage de la papille optique, retard ou défaut de remplissage de la choroïde péripapillaire (ischémie segmentaire) sont caractéristiques.
Dans la vascularite, l’épaississement de la paroi vasculaire et la formation de thrombus entraînent une nécrose ischémique. La vascularite des artères ciliaires courtes postérieures (SPCAs) provoque une ischémie de la partie antérieure du nerf optique, accompagnée d’une ischémie segmentaire de la choroïde.
Les mécanismes oculaires directs sont les suivants :
Inflammation des artères ciliaires courtes postérieures → épaississement pariétal → rétrécissement de la lumière → formation de thrombus → ischémie de la papille optique.
L’occlusion de l’SPCA médiale est la plus fréquente (particulièrement touchée dans 20% des cas).
Les SPCAs vascularisent la lame criblée antérieure et la lame criblée, et assurent également la circulation choroïdienne péripapillaire.
Les cellules dendritiques de la paroi vasculaire agissent comme facteur contributif principal. Les macrophages et les lymphocytes T pénètrent par les vasa vasorum de l’adventice, initiant une cascade pathogène et provoquant une vascularite granulomateuse touchant les artères de moyen et gros calibre.
Âge : principal facteur de risque. Âge médian 75 ans. L’apparition avant 50 ans est extrêmement rare.
Sexe : risque 2 à 6 fois plus élevé chez les femmes.
Ethnie : plus fréquent chez les Blancs d’origine nordique. Rare chez les Noirs et les Asiatiques.
Facteurs génétiques : HLA-DRB1*04, DRW6, DR3 sont associés à une susceptibilité accrue. Les polymorphismes du gène TNF-α et du promoteur de l’IL-10 sont également corrélés à un risque accru. Des cas familiaux d’ACG chez les non-Blancs ont été rapportés 7).
Facteurs environnementaux et infectieux : le virus varicelle-zona (VZV), Chlamydia pneumoniae et le parvovirus B19 sont suspectés.
Mécanismes liés à l’âge : la calcification de la limitante élastique interne, de l’élastine et de la matrice extracellulaire pourrait expliquer l’expression spécifique à l’âge.
Tabagisme, faible IMC, ménopause précoce : tous rapportés comme facteurs de risque.
Association avec la PPR : l’ACG et la PPR montrent une forte corrélation.
COVID-19 : une augmentation de 70 % de l’incidence de l’ACG a été rapportée pendant la pandémie 2). Le SARS-CoV-2 a une affinité pour l’endothélium vasculaire, et la similitude avec la vascularite suggère un lien pathologique.
Le diagnostic de la NOIAA est mené en parallèle avec le diagnostic confirmé d’artérite à cellules géantes. La distinction entre NOIAA et NOIAN étant directement liée à la stratégie thérapeutique, une évaluation rapide et systématique est importante.
VS : sensibilité 86 %. Peut atteindre 70 à 120 mm/h. Valeurs normales : hommes = âge/2, femmes = (âge+10)/2. Cependant, jusqu’à 10 % des cas présentent des valeurs normales.
CRP : sensibilité 97,5 %. Plus spécifique que la VS. Dans les critères de classification ACR/EULAR 2022, une CRP ≥ 10 mg/L a été ajoutée comme élément supplémentaire 4).
VS + CRP combinées : sensibilité 99,2 %, spécificité 97 %. Les protéines de phase aiguë sont élevées dans plus de 80 % des cas.
Autres : la combinaison thrombocytose, CRP et plaquettes a la plus grande utilité diagnostique (p < 0,001) 4).
Biopsie de l'artère temporale (BAT)
Rôle : Gold standard pour le diagnostic définitif de l’artérite à cellules géantes. Réalisée correctement, sensibilité et spécificité > 95 %.
Résultats positifs : épaississement intimal, rupture de la limitante élastique interne, infiltrat inflammatoire chronique avec cellules géantes. Le diagnostic pathologique nécessite une destruction de la limitante élastique interne et un infiltrat de cellules inflammatoires (phase aiguë) ou une fibrose (phase chronique). Les cellules géantes ne sont pas indispensables au diagnostic définitif.
Faux négatifs : taux de faux négatifs de 3 à 5 % dû aux lésions en saut (skip lesions). Certains rapports mentionnent jusqu’à 61 % 6). Un TAB négatif n’exclut pas une GCA.
Moment de la réalisation : la biopsie doit être effectuée dans les jours suivant le début du traitement par stéroïdes.
Imagerie diagnostique
Échographie de l’artère temporale (CDUS) : examen non invasif et reproductible. Sensibilité 77 %, spécificité 96 % 4). Signe du halo (anneau hypoéchogène dû à l’épaississement de la paroi vasculaire), signe de compression, sténose et occlusion sont des signes caractéristiques. En raison des lésions en saut, une exploration bilatérale et multi-territoire est importante 4). Si le signe du halo est positif des deux côtés, la spécificité atteint 100 % 4).
TEP-TDM : peut détecter une accumulation anormale dans l’aorte et ses branches dans la GCA des gros vaisseaux (LV-GCA). L’étude GAPS a montré une sensibilité de 92 % et une spécificité de 85 % 6).
IRM : utile pour différencier AAION et NAION. Vérifier le rehaussement de la gaine du nerf optique et de la graisse orbitaire (central bright spot).
Examens ophtalmologiques
Angiographie à la fluorescéine (FA) : Retard de remplissage de la tête du nerf optique, retard/défaut de remplissage de la choroïde péripapillaire (ischémie segmentaire) sont caractéristiques de l’AAION. Peut être observé avant l’apparition de l’œdème papillaire. Point de différenciation important avec la NAION.
OCT/OCTA : Utile pour évaluer l’œdème papillaire segmentaire, l’épaisseur de la couche des fibres nerveuses rétiniennes (RNFL) et l’état ischémique de la tête du nerf optique.
Critères de classification ACR (1990) et critères ACR/EULAR 2022
Il faut différencier la GCA des autres vascularites telles que la polyartérite noueuse, la granulomatose avec polyangéite (Wegener) et le LED. Un point important est que la GCA n’affecte pas les poumons ni les reins. La syphilis oculaire peut présenter des symptômes similaires à la GCA 10). Il faut également être attentif à la GCA occulte (environ 20 %) qui ne présente pas de symptômes généraux.
QUne biopsie de l'artère temporale négative peut-elle exclure une artérite à cellules géantes ?
A
Non. Les lésions sautées (inflammation présente uniquement dans une partie du vaisseau) entraînent un taux de faux négatifs de 3 à 5 % (certains rapports indiquent jusqu’à 61 % 6)). Même si la TAB est négative, en cas de suspicion clinique de GCA avec élévation de la VS et de la CRP, le traitement doit être poursuivi. Il est important d’évaluer l’ensemble des données cliniques, biologiques et échographiques.
Dès qu’une déficience visuelle est suspectée, le traitement doit être instauré rapidement sans attendre la confirmation par biopsie. L’objectif principal du traitement est de prévenir l’atteinte de l’autre œil ; l’amélioration de l’acuité visuelle de l’œil atteint est rarement obtenue. Seulement 15 à 20 % des patients voient leur vision s’améliorer. Un traitement hospitalier par perfusion massive de stéroïdes est souhaitable.
Traitement par stéroïdes (phase aiguë, phase d’entretien, diminution progressive)
Phase aiguë : Administrer immédiatement de la méthylprednisolone par voie intraveineuse à raison de 1 g/jour pendant 3 à 5 jours.
Phase d’entretien : Passer à la prednisolone par voie orale à raison de 1 mg/kg/jour.
Diminution progressive : Réduire lentement la dose sur au moins 4 à 6 mois en fonction de l’état général et de la VS. Dans certains cas, cela peut prendre plus d’un an.
Attention : L’administration de stéroïdes un jour sur deux n’est pas recommandée.
En l’absence de symptômes oculaires ou du système nerveux central, le traitement peut débuter par 30 à 40 mg/jour de prednisolone.
Envisager en cas d’effets indésirables liés à l’administration prolongée de corticoïdes (syndrome cushingoïde, hyperglycémie, ostéoporose, symptômes gastro-intestinaux, etc., survenant chez environ 60 % des patients).
Tocilizumab (tocilizumab ; inhibiteur du récepteur de l’IL-6) : approuvé par la FDA en 2017 pour le traitement de l’artérite à cellules géantes (ACG). Les essais contrôlés randomisés (ECR) ont démontré son effet d’épargne des corticoïdes et son efficacité pour obtenir une rémission sur 12 mois4). Des rapports indiquent son efficacité dans la neuropathie optique ischémique antérieure aiguë (NOIAA) résistante aux stéroïdes. L’utilisation de TCZ 162 mg en injection sous-cutanée a également été rapportée dans des cas d’ACG post-vaccin COVID-198)9).
Méthotrexate : augmente le taux d’arrêt durable des corticoïdes et réduit le risque de rechute. Dans les rapports de cas, une dose de 15 mg/semaine est utilisée en association1). En cas de rechute, une augmentation de la dose de corticoïdes associée au MTX peut être recommandée.
Aspirine à faible dose : peut être envisagée en association pour la prévention des complications cardiaques et ischémiques cérébrovasculaires.
QLa vision peut-elle être restaurée par un traitement aux stéroïdes ?
A
L’amélioration de la vision de l’œil atteint est rarement attendue. Seulement 15 à 20 % des patients voient leur vision s’améliorer, et dans la plupart des cas, une baisse de vision persiste. L’objectif principal du traitement aux stéroïdes est de prévenir l’atteinte de l’œil controlatéral.
La physiopathologie de l’artérite à cellules géantes repose sur deux mécanismes immunitaires principaux. Il s’agit d’une vascularite granulomateuse à médiation T, initiée par l’activation des cellules dendritiques de la paroi vasculaire, qui affecte sélectivement les artères de moyen et gros calibre.
Cascade immunitaire et inflammatoire (modèle à deux axes)
Réponse immunitaire innée médiée par l’IL-6 : les macrophages circulants, les neutrophiles et les monocytes produisent de l’IL-6.
Hyperactivation de la réponse de phase aiguë : corrélée à l’augmentation de la CRP, de l’haptoglobine, du fibrinogène et du complément.
Cause des symptômes systémiques : provoque fièvre, fatigue, perte de poids, etc.
Thérapie ciblée : l’axe IL-6–Th17–IL-17/IL-21 peut être inhibé par les glucocorticoïdes et le tocilizumab (inhibiteur de l’IL-6).
Réponse antigène-spécifique
Pénétration dans la paroi artérielle : les macrophages et les lymphocytes T pénètrent via les vasa vasorum de l’adventice.
Cascade immunitaire : activation des lymphocytes T → recrutement des lymphocytes T CD4+ → polarisation Th1/Th17 → production d’IFN-γ/IL-17 → recrutement des monocytes → différenciation en macrophages → formation de cellules géantes.
Destruction de la paroi vasculaire : destruction de la limitante élastique interne par les métalloprotéases et les espèces réactives de l’oxygène.
Non-implication des lymphocytes B : l’implication des lymphocytes B n’a pas été confirmée, ce qui constitue un point de différenciation important avec les vascularites associées aux ANCA.
L’apport sanguin au nerf optique est principalement assuré par les artères ciliaires postérieures courtes (SPCAs) et les branches de l’artère centrale de la rétine.
Les SPCAs irriguent la région prélamaire et laminaire, et participent également à la circulation choroïdienne péripapillaire.
Dans l’artérite à cellules géantes (GCA), l’occlusion thrombotique des SPCAs (particulièrement touchées dans 20 % des cas) provoque une ischémie de la tête du nerf optique.
Les examens post-mortem de la neuropathie optique ischémique antérieure aiguë (AAION) ont confirmé un œdème de la tête du nerf optique avec nécrose des régions prélamaire, laminaire et rétrolaminaire, ainsi qu’un infiltrat inflammatoire chronique.
La rupture de la lame élastique interne est caractéristique, et les cellules géantes sont situées à proximité de la lame élastique interne rompue. La phase aiguë est dominée par une infiltration lymphocytaire, tandis que la phase chronique entraîne une fibrose. En réaction de guérison à l’inflammation, un épaississement de l’intima, une prolifération de myofibroblastes et un dépôt de matrice extracellulaire se produisent, conduisant à une sténose et une occlusion vasculaires.
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
L’upadacitinib, un inhibiteur sélectif oral de JAK1, a été approuvé par la FDA en 2025 pour le traitement de l’artérite à cellules géantes (ACG). Il est considéré comme une nouvelle option thérapeutique ciblant la voie IL-6–JAK–STAT.
Preuves d’essais contrôlés randomisés (ECR) pour le tocilizumab
Le tocilizumab a démontré dans des ECR un effet d’épargne des glucocorticoïdes (GC) et une efficacité pour atteindre la rémission sur 12 mois 4), s’établissant comme une alternative thérapeutique réduisant la toxicité associée à l’administration prolongée de GC.
Fast-track pathway (voie de diagnostic rapide par échographie)
La voie de diagnostic rapide de l’artérite à cellules géantes utilisant l’échographie se répand en Europe. L’introduction de la fast-track pathway a montré une réduction de la perte de vision, une limitation du surtraitement et une amélioration du rapport coût-efficacité 4). L’échographie est non invasive, reproductible et permet d’évaluer plusieurs territoires artériels en une seule fois, ce qui en fait un outil majeur pour le diagnostic précoce.
Un sous-type d’ACG survenant dans le contexte d’un syndrome myélodysplasique (SMD) (GCA-MDS) est reconnu. On estime que 10 à 20 % des patients atteints de SMD développent une maladie auto-immune. La GCA-MDS pourrait avoir une prévalence plus faible de symptômes classiques (céphalées, claudication de la mâchoire, AAION, etc.) que l’ACG typique, et tend à être plus corticodépendante, avec une survie sans stéroïdes et sans rechute réduite. L’ajout d’agents hypométhylants (azacitidine/décitabine) pourrait être bénéfique, et une étude prospective (NCT02985190) est en cours. Le plus grand rapport est une série française multicentrique de 21 cas en 2019.
Une augmentation de 70 % de l’incidence de l’ACG pendant la pandémie de COVID-19 a été rapportée 2). Plusieurs rapports indiquent une augmentation des cas d’ACG en 2020, avec une hausse du taux de complications oculaires, et suggèrent une implication des lésions endothéliales, de l’immunité Th1 et du système monocyte-macrophage 2). Des rapports de cas suggèrent également que le SARS-CoV-2 pourrait avoir déclenché l’ACG.
Plusieurs cas de GCA après vaccination contre la COVID-19 ont été rapportés. Yoshimoto et al. (2023) ont examiné 14 cas et ont rapporté que la période entre l’apparition et le diagnostic variait de 2 semaines à 4 mois (moyenne d’environ 6 semaines) 8). Une cécité est survenue dans 2 des 14 cas.
Sverdlichenko et al. (2022) ont rapporté une association avec le syndrome de Horner (ptosis partiel et myosis) chez 2 des 53 patients atteints de GCA 5). Le mécanisme présumé est une ischémie des premiers neurones sympathiques dans le tronc cérébral due à une vascularite de l’artère vertébrale et de ses branches. Pour un nouveau syndrome de Horner chez les personnes de plus de 50 ans, il est recommandé de vérifier les symptômes de GCA et de doser les marqueurs inflammatoires.
La GCA peut provoquer diverses complications ischémiques en dehors de la vision 3).
Accident vasculaire cérébral : survient dans 2 à 7 % des cas.
Nécrose de la langue et du cuir chevelu : complications rares mais graves.
Complications artérielles périphériques.
Syndrome de Charles Bonnet : hallucinations visuelles chroniques survenant après une perte de vision permanente. Rapporté chez 0,4 à 30 % des patients malvoyants 3).
Hayreh et al. (2021) ont rapporté un cas de 3 frères sur 5 d’origine indienne ayant développé une artérite à cellules géantes, suggérant un mode de transmission autosomique récessif 7). Ce cas est considéré comme le premier rapport de GCA familiale chez des non-Blancs.
Szydełko-Paśko U, Przeździecka-Dołyk J, Kręcicka J, Małecki R, Misiuk-Hojło M, Turno-Kręcicka A. Arteritic Anterior Ischemic Optic Neuropathy in the Course of Giant Cell Arteritis After COVID-19. The American journal of case reports. 2022;23:e933471. doi:10.12659/AJCR.933471. PMID:35015754; PMCID:PMC8762612.
Jalaledin DS, Ross C, Makhzoum JP. Rare Ischemic Complications of Giant Cell Arteritis: Case Series and Literature Review. The American journal of case reports. 2022;23:e937565. doi:10.12659/AJCR.937565. PMID:36240129; PMCID:PMC9578098.
Piccus R, Hansen MS, Hamann S, Mollan SP. An update on the clinical approach to giant cell arteritis. Clinical medicine (London, England). 2022;22(2):107-111. doi:10.7861/clinmed.2022-0041. PMID:35304369; PMCID:PMC8966809.
Sverdlichenko I, Lam C, Donaldson L, Margolin E. Horner Syndrome in Giant Cell Arteritis: Case Series and Review of the Literature. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2022;42(3):340-345. doi:10.1097/WNO.0000000000001593. PMID:35427255.
Stewart C, Asif RH, Dakkak T, Singh H, Javaid MA, Patel N. Diagnostic Dilemmas in Giant Cell Arteritis: Overcoming Anchoring Bias. Case reports in rheumatology. 2025;2025:6632374. doi:10.1155/crrh/6632374. PMID:40726518; PMCID:PMC12303627.
Yoshimoto K, Kaneda S, Asada M, Taguchi H, Kawashima H, Yoneima R, Matsuoka H, Tsushima E, et al. Giant Cell Arteritis after COVID-19 Vaccination with Long-Term Follow-Up: A Case Report and Review of the Literature. Medicina (Kaunas, Lithuania). 2023;59(12). doi:10.3390/medicina59122127. PMID:38138230; PMCID:PMC10744572.
Wakabayashi H, Iwayanagi M, Sakai D, et al. Development of giant cell arteritis after vaccination against SARS-CoV2. Medicine. 2023;102(21):e33814.
Qadir A, Khakwani AS, Khan MR, et al. Ocular Syphilis Mimicking Giant Cell Arteritis. Cureus. 2022;14(9):e29286.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.