โรคเผือก (Albinism) เป็นกลุ่มโรคทางพันธุกรรมที่มีลักษณะขาดหรือลดลงของเมลานิน ร่วมกับภาวะโฟเวียเจริญไม่เต็มที่ (foveal hypoplasia) อาตา (nystagmus) และการไขว้ของเส้นทางเดินภาพผิดปกติ
การจำแนกหลักมี 3 ประเภท: โรคเผือก ชนิดผิวหนังและตา (OCA) โรคเผือก ชนิดตา (OA) และโรคเผือก ที่เป็นส่วนหนึ่งของกลุ่มอาการ (เช่น กลุ่มอาการเฮอร์แมนสกี-พุดลัก)
ภาวะโฟเวียเจริญไม่เต็มที่เป็นปัจจัยสำคัญที่สุดที่ทำให้การมองเห็น ไม่ดี ไม่มีการรักษาให้หายขาด และการทำงานของการมองเห็น คงที่
การตรวจพบการไขว้ของเส้นทางเดินภาพผิดปกติด้วย VEP มีความสำคัญอย่างยิ่งในการวินิจฉัย
ในญี่ปุ่น โรคนี้ถูกกำหนดให้เป็นโรคหายากเฉพาะ และการรักษามุ่งเน้นที่การดูแลการมองเห็น เลือนรางเฉพาะทางและการให้คำปรึกษาทางพันธุกรรม
ในชนิดที่เป็นกลุ่มอาการ (HPS และ CHS ) มีภาวะแทรกซ้อนทั่วร่างกายที่ปอด ระบบทางเดินอาหาร และเลือด จำเป็นต้องทำงานร่วมกันหลายสาขา
โรคเผือก (Albinism) เป็นกลุ่มโรคทางพันธุกรรมที่เกิดจากการกลายพันธุ์ของยีนที่เกี่ยวข้องกับการสังเคราะห์หรือการขนส่งเม็ดสีเมลานิน ส่งผลให้ขาดหรือลดลงของเม็ดสีที่ผิวหนัง ผม และดวงตา นอกจากภาวะขาดเม็ดสีแล้ว โรคนี้ยังมีลักษณะความผิดปกติของตาและเส้นทางเดินภาพที่จำเพาะ เช่น ภาวะโฟเวียเจริญไม่เต็มที่ อาตา และการไขว้ของเส้นทางเดินภาพผิดปกติ
ไม่มีการรักษาให้หายขาด และความบกพร่องทางการมองเห็น ไม่ดำเนินไป (คงที่) ในญี่ปุ่น โรคนี้ถูกกำหนดให้เป็นโรคหายากเฉพาะ (ตามกฎหมายโรคหายาก)
โรคเผือก แบ่งออกเป็น 3 กลุ่มหลัก
โรคเผือกชนิดผิวหนังและตา (OCA)
ถ่ายทอดทางพันธุกรรมแบบด้อยบนออโตโซม : กลุ่มที่พบบ่อยที่สุด ทำให้เกิดการขาดเม็ดสีที่ผิวหนัง ผม และดวงตา
8 ยีนและ 8 ชนิดย่อย : OCA1 (TYR), OCA2 (ยีน OCA2/P), OCA3 (TYRP 1), OCA4 (SLC45A2), OCA8 (DCT) และอื่นๆ
ความชุก : ประมาณ 1/17,000 ทั่วโลก ในคนผิวขาว OCA1 คิดเป็นประมาณ 50% ในคนแอฟริกัน OCA2 พบมากที่สุด (1/10,000)1) ในชาวจีนฮั่น OCA1 คิดเป็น 70.1% และ OCA2 คิดเป็น 10.2%1)
โรคเผือกชนิดตา (OA)
ถ่ายทอดทางพันธุกรรมแบบด้อยบนโครโมโซม X : เกิดในเพศชาย เม็ดสีที่ผิวหนังและผมปกติหรือลดลงเล็กน้อย โดยมีอาการทางตาเป็นหลัก
การกลายพันธุ์ของยีน GP R143 : เกี่ยวข้องกับการส่งสัญญาณการสร้างเมลาโนโซม มีรายงานการกลายพันธุ์ 192 ชนิด3)
หญิงที่เป็นพาหะ : แสดงรูปแบบเม็ดสีต่ำแบบโมเสกคล้ายรอยเปื้อนโคลนที่จอตา ประมาณ 80% มีความผิดปกติของเม็ดสีจอประสาทตา 3)
โรคเผือกแบบกลุ่มอาการ
กลุ่มอาการเฮอร์แมนสกี-พัดลัก (HPS) : OCA + ความผิดปกติของเกล็ดเลือด (แนวโน้มเลือดออก) + ความผิดปกติของการสร้าง LRO มี 11 ชนิดย่อย4, 5) อาจมีภาวะพังผืดในปอดและโรคลำไส้อักเสบร่วมด้วย
กลุ่มอาการเชเดียก-ฮิงาชิ (CHS ) : OCA + ภูมิคุ้มกันบกพร่อง + ความผิดปกติทางระบบประสาท การกลายพันธุ์ของยีน LYST
12 ยีน : มีการระบุยีน 12 ชนิดสำหรับโรคเผือก แบบกลุ่มอาการ2)
ความชุกทั่วโลกประมาณ 1/17,0001) และในยุโรปรายงานประมาณ 1/12,0002) ในคนญี่ปุ่น การกระจายที่ทราบคือ OCA1 34%, OCA4 27%, HPS1 10%
Q
โรคเผือกเป็นกรรมพันธุ์หรือไม่? สามารถเกิดโรคได้แม้ว่าไม่มีใครในครอบครัวเป็นโรคเผือกหรือไม่?
A
OCA เป็นโรคถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อย โดยหากพ่อและแม่เป็นพาหะมียีนกลายพันธุ์คนละ 1 อัลลีล โอกาสที่ลูกจะเป็นโรคคือ 25% โรคนี้สามารถเกิดขึ้นได้แม้พ่อแม่มีลักษณะปกติ ดังนั้นจึงไม่ใช่เรื่องแปลกแม้ไม่มีประวัติครอบครัว OA เป็นโรคถ่ายทอดแบบ X-linked ด้อย ซึ่งส่วนใหญ่เกิดในเพศชาย โดยมารดาเป็นพาหะ การตรวจทางพันธุกรรมและการให้คำปรึกษาทางพันธุกรรม มีความสำคัญ
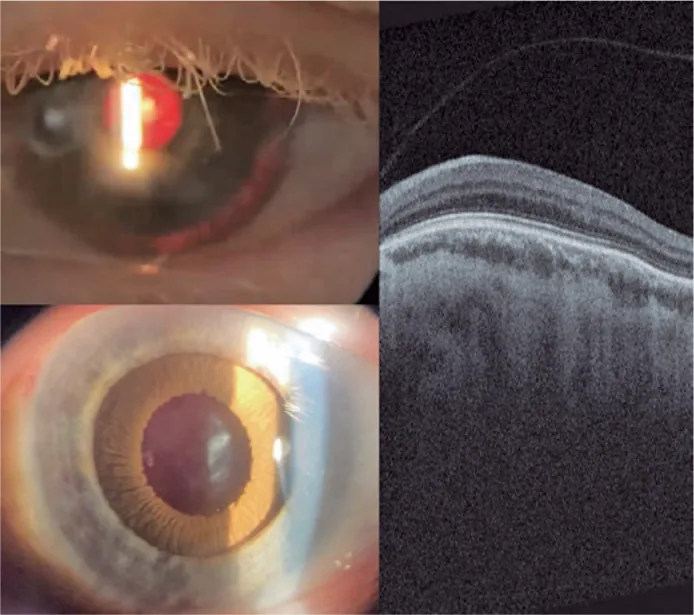
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PM
CI D: PMC11620172. License: CC BY.
ภาพถ่ายด้วยหลอดกรีดตาซ้าย 30 วันหลังผ่าตัด แสดง
ม่านตาเทียม บนซัลคัสซิลิอารีผ่านการส่องสว่างย้อนกลับและหลังการขยาย
ม่านตา ด้วยยา
OCT จอประสาทตา แสดงการไม่มีหลุมฟอเวียและการคงอยู่ของชั้นใน
จอประสาทตา บริเวณที่คาดว่าเป็นฟอเวีย (ภาวะ macular hypoplasia)
ด้านล่างนี้คือค่าประมาณการมองเห็น ตามชนิดย่อยหลัก
ชนิดย่อย ช่วงการมองเห็น โดยประมาณ OCA1 มัก ≤ 0.1 OCA2/OCA4 0.1 ถึง 0.3 OCA8 0.1 ถึง 0.4 (LogMAR) 2) OA1 0.1 ถึง 0.4 HPS-11 ประมาณ 20/200 5)
การมองเห็น ไม่ดีกลัวแสง (อาการเคืองตา) : การขาดเม็ดสีในม่านตา ทำให้แสงกระจาย เกิดอาการกลัวแสง รุนแรงความผิดปกติของการหักเหแสง : มักพบความผิดปกติของการหักเหแสงระดับรุนแรง (สายตาสั้น สายตายาว สายตาเอียง )อาตา (ตากระตุก )อาตา แบบลูกตุ้มที่ปรากฏเมื่ออายุ 2-3 เดือน
ภาวะโฟเวียเจริญไม่เต็มที่ : ปัจจัยสำคัญที่สุดที่ทำให้การมองเห็น ไม่ดี การไม่มีรอยบุ๋มโฟเวียยืนยันโดย OCT ในการถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน มักไม่เห็นบริเวณไร้หลอดเลือดรอบโฟเวีย ใน OCA8 ทุกรายมีระดับ 3 และระดับมีความสัมพันธ์กับความคมชัดของการมองเห็น 2) การส่องผ่านของม่านตา : เนื่องจากการขาดเม็ดสีม่านตา จึงพบการส่องผ่านเพิ่มขึ้นเมื่อตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด ใน OCA8 ทุกรายมีระดับ 3 2) อาตา (INS)อาตา แบบสลับข้างเป็นระยะไม่สมมาตรพบในประมาณ 10% ของ INS ทั้งหมด และสูงถึง 37% ใน INS จากโรคเผือก 2) การไขว้เส้นทางเดินประสาทตาผิดปกติ (chiasmal misrouting) : เส้นใยจอประสาทตา ส่วนขมับ ซึ่งปกติควรเดินทางแบบ ipsilateral (ไม่ไขว้) กลับไขว้ไปยังด้านตรงข้ามมากเกินไปที่จุดไขว้ประสาทตา ตรวจพบโดย VEP ใน OCA8 มีรายงานค่าสัมประสิทธิ์จุดไขว้ -0.97/-0.90 แสดงถึงความไม่สมมาตรอย่างรุนแรง 2) ตาเหล่ 2) จอตาสีจาง : เนื่องจากการขาดเม็ดสีของเยื่อบุผิวจอตา ทำให้มองเห็นหลอดเลือดคอรอยด์ จอตาของพาหะ OA1 : 94% มีการเปลี่ยนแปลงของเม็ดสีเป็นปื้นด้านหน้าหลอดเลือดรูปโค้ง 74% มีการส่องผ่านของม่านตา 3) อาจพบจุดสีจางในจอตาส่วนปลาย (จอตาแบบโมเสก) ซึ่งมีประโยชน์ในการระบุพาหะ
Q
ความคมชัดของการมองเห็นในโรคเผือกเปลี่ยนแปลงตลอดชีวิตหรือไม่?
A
ความบกพร่องทางการมองเห็น จากโรคเผือก เป็นแบบคงที่ (ไม่ลุกลาม) และไม่แย่ลงตามอายุ อย่างไรก็ตาม หากไม่ได้รับการแก้ไขค่าสายตาที่เหมาะสมหรือการรักษาภาวะตาขี้เกียจ การมองเห็น อาจยังคงแย่โดยไม่ดีขึ้น การแทรกแซงตั้งแต่เนิ่นๆ ด้วยการแก้ไขค่าสายตาและการดูแลผู้มีความบกพร่องทางการเห็นเป็นสิ่งสำคัญ
ด้านล่างนี้คือยีนก่อโรคหลักและลักษณะเฉพาะของแต่ละชนิดย่อย
ชนิดย่อย ยีนก่อโรค ลักษณะสำคัญ OCA1 TYR การขาดไทโรซิเนส OCA2 OCA2 (ยีน P) การควบคุม pH ของเมลาโนโซม1) OCA3 TYRP 1 การสังเคราะห์ยูเมลานิน OCA4 SLC45A2 ตัวขนส่งเมลาโนโซม OCA8 DCT (TYRP 2) การเปลี่ยนโดปาโครม 2) OA1 GP R143สัญญาณเมลาโนโซม 3) HPS1/HPS4 HPS1/HPS4 คอมเพล็กซ์ BLOC-3 4) HPS11 BLOC1S5 คอมเพล็กซ์ BLOC-1 5)
การทำงานของโปรตีน OCA2 : อยู่ในตระกูล antiporter Na⁺/H⁺ ที่มีโครงสร้างเกลียว α ผ่านเยื่อหุ้ม 12 ครั้ง 1) ในฐานะองค์ประกอบของช่องไอออนลบจำเพาะของเมลาโนโซม ควบคุม pH ของเมลาโนโซมระยะ I/II โดยการควบคุมกระแสคลอไรด์ 1) มีการกลายพันธุ์ที่ก่อโรค 477 รายการที่ลงทะเบียนใน ClinVar 1)
DCT (TYRP 2) : เร่งปฏิกิริยาการเปลี่ยนโดปาโครมเป็น DHICA (กรด 5,6-ไดไฮดรอกซีอินโดล-2-คาร์บอกซิลิก) 2) เป็นยีนที่ก่อให้เกิด OCA8
GP R143 (OA1)3)
HPS (คอมเพล็กซ์ BLOC) : HPS1/HPS4 ทำหน้าที่เป็นคอมเพล็กซ์ BLOC-3 ในฐานะ GEF (ปัจจัยแลกเปลี่ยนนิวคลีโอไทด์กัวนีน) สำหรับ Rab32/38 4) BLOC-1 ประกอบด้วย 8 หน่วยย่อยและเกี่ยวข้องกับการรีไซเคิลเอนโดโซม และการกลายพันธุ์ของยีน BLOC1S5 (BLOS3) ทำให้เกิด HPS-11 (อธิบายครั้งแรกในปี 2020) 5)
การแต่งงานในเครือญาติ : ความชุกเพิ่มขึ้นในประชากรที่มีอัตราการแต่งงานในเครือญาติสูง เช่น ปากีสถาน 4) ความแตกต่างทางเชื้อชาติและภูมิภาค : ความถี่สูงในบางพื้นที่ของแอฟริกาการกลายพันธุ์แบบ compound heterozygous : มีการระบุการกลายพันธุ์แบบ compound heterozygous ใหม่ใน OCA2 ซึ่งมีส่วนทำให้เกิดความหลากหลายของฟีโนไทป์ 1) แบบสามอัลลีล (OCA1) : การรวมกันของสามอัลลีลของยีน TYR อาจทำให้เกิด OCA ระดับเล็กน้อย 2)
Q
เหตุใดการรู้ชนิดของโรคเผือกจึงมีความสำคัญ?
A
เนื่องจากการพยากรณ์การมองเห็น การมีภาวะแทรกซ้อนทางระบบ และรูปแบบการถ่ายทอดทางพันธุกรรมแตกต่างกันตามชนิด การวินิจฉัยชนิดย่อยที่แม่นยำจึงมีความสำคัญ โดยเฉพาะ HPS มักมีแนวโน้มเลือดออกง่าย พังผืดในปอด และโรคลำไส้อักเสบ จึงต้องระมัดระวังก่อนการผ่าตัดหรือถอนฟัน การระบุชนิดย่อยด้วยการตรวจทางพันธุกรรมเชื่อมโยงโดยตรงกับการวางแผนการจัดการที่เหมาะสม
OCA ทั่วไปสามารถวินิจฉัยทางคลินิกได้จากผมขาว ผิวขาว และอาการทางตา (ม่านตา โปร่งแสง อาตา ) อย่างไรก็ตาม ใน OA1 ของญี่ปุ่น เม็ดสียังคงเหลืออยู่ในม่านตา ดังนั้นหากผู้ป่วยมาด้วยอาการทางตาเพียงอย่างเดียว การวินิจฉัยอาจล่าช้า
กล้องจุลทรรศน์ชนิดกรีด (Slit-lamp) : ยืนยันการโปร่งแสงของม่านตา ประเมินระดับความรุนแรงOCT (เครื่องตรวจชั้นจอประสาทตา )6) VEP (ศักย์ไฟฟ้าจากการกระตุ้นการมองเห็น )VEP 3 ขั้วเพื่อประเมินความไม่สมมาตรของคลื่นระหว่างสมองซีก 2) วัดปริมาณด้วยค่าสัมประสิทธิ์ไคแอสมาทิก (optic chiasm coefficient) (-1 = ไม่สมมาตรสูงสุด, +1 = ปกติ) 2) การวัดมุมแลมบ์ดา : มุมแลมบ์ดามากกว่า 5 องศาเป็นตัวบ่งชี้ทางคลินิกที่ชัดเจนของภาวะเผือก 2) การถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน (FA ) : ทำให้เห็นรูปแบบโมเสกของจอประสาทตา ในพาหะของ OA1 ชัดเจนขึ้นกล้องจุลทรรศน์อิเล็กตรอนของเกล็ดเลือด : ใช้ในการวินิจฉัย HPS ที่แน่นอน การไม่มี dense body เป็นลักษณะเฉพาะ 4) การตรวจทางพันธุกรรม : การหาลำดับเอ็กโซมทั้งหมด (WES) 1) หรือการหาลำดับแบบแพเนล 2, 4, 5) สำคัญเป็นพิเศษเมื่อฟีโนไทป์ไม่รุนแรงหรือผิดปกติ
ไม่มีการรักษาที่หายขาด เป้าหมายของการรักษาคือการเพิ่มประสิทธิภาพการมองเห็น สูงสุดและจัดการภาวะแทรกซ้อน
การจัดการด้านการมองเห็น
การแก้ไขค่าสายตา : แว่นตาตั้งแต่วัยทารกเป็นพื้นฐาน การแทรกแซงตั้งแต่เนิ่นๆ เพื่อป้องกันภาวะตาขี้เกียจ เป็นสิ่งสำคัญ
แว่นตาป้องกันแสง : ใช้เลนส์ที่บังแสงประมาณ 20% ในร่ม และประมาณ 80% กลางแจ้ง 5)
คอนแทคเลนส์สี ม่านตา อาการกลัวแสง
การดูแลผู้มีสายตาเลือนราง
อาตาและตาเหล่
การผ่าตัดตาเหล่ : ดำเนินการเพื่อความสวยงาม บางครั้งอาจทำการผ่าตัด Dawson-Trick-Litzkow (DTL) เพื่อลดอาตา
การรักษาอาตา : ไม่มีการรักษาที่หายขาด การผ่าตัดตาเหล่ ถูกพิจารณาเพื่อจัดการกับท่าศีรษะที่ผิดปกติ (การรักษา null zone)
การจัดการผิวหนังและระบบร่างกาย
การป้องกันแสงแดด : ครีมกันแดดที่ป้องกันรังสี UVB เสื้อผ้า และหมวกเพื่อปกป้องผิว ควรระวังความเสี่ยงของมะเร็งเซลล์สความัส แม้ในคนญี่ปุ่น
การจัดการ HPS : ยา NSAID (รวมถึงแอสไพริน) โดยหลักการแล้วห้ามใช้เนื่องจากทำให้ความผิดปกติของเกล็ดเลือดแย่ลง แนะนำให้ติดตามการทำงานของปอดเป็นประจำตั้งแต่วัยรุ่น 4)
การให้คำปรึกษาทางพันธุกรรม
สำหรับพังผืดในปอดที่เกิดร่วมกับ HPS จะใช้ยาต้านการเกิดพังผืด
Liu และคณะ (2025) รายงานผู้ป่วย HPS ที่มีการกลายพันธุ์แบบโฮโมไซกัสใหม่ใน HPS4 ซึ่งพังผืดในปอดคงที่เป็นเวลา 18 เดือนหลังการให้ยา nintedanib 4) Pirfenidone ก็เป็นทางเลือกในการรักษาที่ใช้ในลักษณะเดียวกัน
ในผู้ป่วย HPS ก่อนการผ่าตัด การคลอดบุตร หรือการถอนฟัน ควรประสานงานกับแผนกโลหิตวิทยาเพื่อประเมินความเสี่ยงต่อการตกเลือด ยา NSAIDs อาจทำให้การทำงานของเกล็ดเลือดแย่ลงและควรใช้ด้วยความระมัดระวัง
แว่นตาป้องกันแสงช่วยลดอาการกลัวแสง แต่ไม่ได้ช่วยให้การมองเห็น ดีขึ้น
ความบกพร่องทางการมองเห็น ในโรคเผือก เป็นแบบคงที่ และปัจจุบันยังไม่มีการผ่าตัดหรือยาที่พิสูจน์แล้วว่าช่วยปรับปรุงการมองเห็น
Q
การบำบัดด้วยยีนสำหรับโรคเผือกได้ถูกนำมาใช้ในทางปฏิบัติหรือยัง?
A
ปัจจุบัน การบำบัดด้วยยีน สำหรับโรคเผือก ยังไม่ได้ถูกนำมาใช้ในทางปฏิบัติ การบำบัดด้วยยีน ได้รับการอนุมัติสำหรับโรคจอประสาทตา ทางพันธุกรรมอื่นๆ เช่น โรคจอประสาทตาฉีกขาด (retinoschisis) และ LCA (ตัวอย่าง: Luxturna) และกำลังมีการวิจัยเพื่อประยุกต์ใช้กับโรคเผือก 6) ปัจจุบัน การแก้ไขค่าสายตาและการดูแลผู้มีความบกพร่องทางการมองเห็น เป็นแกนหลักของการรักษามาตรฐาน
การสังเคราะห์เมลานินเป็นไปตามวิถีต่อไปนี้ภายในเมลาโนโซม:
ไทโรซีน → L-DOPA → DOPA ควิโนน → (ยูเมลานินหรือฟีโอเมลานิน)
การเปลี่ยนโดปาโครมเป็น DHICA (กรด 5,6-ไดไฮดรอกซีอินโดล-2-คาร์บอกซิลิก) ถูกเร่งโดย DCT (TYRP 2) 2) ซึ่งเป็นปฏิกิริยาที่จำเป็นสำหรับการสังเคราะห์ยูเมลานิน
ความผิดปกติของการเจริญเติบโตของเมลาโนโซมใน OCA2 : การทำงานของโปรตีน OCA2 ที่ลดลงขัดขวางการเปลี่ยนผ่านของเมลาโนโซมไปยังระยะที่ 4 (เมลาโนโซมที่เจริญเต็มที่) ทำให้เมลาโนโซมที่ยังไม่เจริญเต็มที่ระยะ I/II เพิ่มขึ้น 1) กลไกหลักคือการควบคุม pH ที่ผิดปกติผ่านการควบคุมกระแสคลอไรด์ 1)
เมื่อการสร้างเม็ดสีของเยื่อบุผิวเม็ดสีจอประสาทตา ไม่เหมาะสม ผลการบังแสงในระหว่างการพัฒนาจะสูญเสียไป ขัดขวางการพัฒนาทางสัณฐานวิทยาของฟอฟีียร์ (การสร้างบริเวณไร้หลอดเลือดและการเคลื่อนย้ายเซลล์รูปกรวย อย่างหนาแน่น) นี่คือสาเหตุหลักของการมองเห็น ไม่ดีในโรคเผือก
ในโรคเผือก เส้นใยจากจอประสาทตา ส่วนขมับจะไขว้ไปยังด้านตรงข้ามมากเกินไปที่ออปติกไคแอซึม ส่งผลให้เส้นใยจำนวนมากถูกประมวลผลเป็นเส้นใยส่วนจมูก การ “เดินทางผิด” นี้สามารถตรวจพบได้จากความไม่สมมาตรของสัมประสิทธิ์ไคแอซึมในการตรวจ VEP 2)
ในการศึกษา OCA8 โดย Rateaux และคณะ (2025) ได้ใช้แบบจำลองหนูที่มีการกลายพันธุ์ DCT ซึ่งทำให้ L-DOPA ลดลงเหลือ 50% ของชนิดปกติ และพบว่าการเสริม L-DOPA สามารถฟื้นฟูความผิดปกติทางจักษุวิทยาได้ 2) ซึ่งบ่งชี้ถึงบทบาทของ L-DOPA ในฐานะสารตัวกลางในการสังเคราะห์เมลานินต่อการสร้างทางเดินภาพ
การกลายพันธุ์ GP R143 (OA1) : การสูญเสียการทำงานของโปรตีน GP R143 ทำให้การขนส่งถุงเมลาโนโซมบกพร่อง นำไปสู่การเกิดเมลาโนโซมขนาดใหญ่ 3)
HPS (ความผิดปกติของกลุ่ม BLOC) : การสูญเสียการทำงานของ BLOC-3 ทำให้การกระตุ้น Rab32/38 บกพร่อง ส่งผลให้การสร้างออร์แกเนลล์ที่เกี่ยวข้องกับเมลาโนโซม (LRO) ล้มเหลวโดยรวม 4) ออร์แกเนลล์หนาแน่นในเกล็ดเลือดและตัวลามิลลาในเซลล์เยื่อบุปอดชนิดที่ 2 ได้รับผลกระทบ การสะสมของสารคล้ายซีรอยด์จากตัวลามิลลาที่ผิดปกติเชื่อมโยงกับพังผืดในปอด 4)
BLOC-1 (HPS-11) : ทำหน้าที่เป็นกลุ่มโปรตีน 8 หน่วยย่อยรวมถึง BLOC1S5 (BLOS3) ควบคุมการรีไซเคิลเอนโดโซม 5)
L-DOPA เป็นสารตัวกลางในการสังเคราะห์เมลานินและมีบทบาทสำคัญในการพัฒนาจอประสาทตา และการสร้างทางเดินภาพ
ในแบบจำลองหนู OCA8 ของ Rateaux และคณะ (2025) การลดลงของ L-DOPA เนื่องจากการกลายพันธุ์ DCT เป็นปัจจัยหลักของฟีโนไทป์ทางจักษุวิทยา และการเสริม L-DOPA ช่วยปรับปรุงความผิดปกติ 2) กำลังมีการศึกษาการประยุกต์ใช้ในมนุษย์ 6)
การประเมินการปรับปรุงการมองเห็น ด้วยการให้เลโวโดปาในผู้ป่วยโรคเผือก กำลังดำเนินการอยู่ โดยคาดหวังผลในการกระตุ้นการพัฒนาจอประสาทตา 6)
นิติซิโนน ซึ่งเป็นสารยับยั้งเอนไซม์ 4-ไฮดรอกซีฟีนิลไพรูเวตไดออกซิจีเนส (4-HPPD) กำลังถูกศึกษาเกี่ยวกับความสามารถในการเพิ่มเมลานินภายในตาโดยการปรับวิถีเมแทบอลิซึมของไทโรซีน 6)
การวิจัยพื้นฐานเกี่ยวกับการบำบัดด้วยการเติมยีนที่กำหนดเป้าหมายไปที่ยีนที่เกี่ยวข้องกับ OCA (TYR, OCA2 ฯลฯ) กำลังดำเนินไป การอนุมัติ Luxturna (การเติมยีน RPE 65) สำหรับ LCA (ภาวะตาบอดแต่กำเนิดชนิด Leber) กำลังเร่งการวิจัยการบำบัดด้วยยีน สำหรับโรคจอประสาทตา ทางพันธุกรรมโดยทั่วไป 6)
Jiang และคณะ (2024) ระบุการกลายพันธุ์แบบ heterozygous แบบผสมใหม่ (c.635A>G/c.2359+1G>T) ในยีน OCA2 โดยการหาลำดับเอ็กโซมทั้งหมด (WES) ในครอบครัวชาวจีน 1) การวิเคราะห์เชิงหน้าที่ยืนยันว่าโปรตีนกลายพันธุ์มีการเคลื่อนย้ายไปยังเมลาโนไซต์ที่บกพร่องเมื่อเทียบกับชนิดปกติ
Boeckelmann และคณะ (2021) ให้รายละเอียดสเปกตรัมทางคลินิกของ HPS-11 จากการกลายพันธุ์ BLOC1S5 ผ่านการศึกษา 5 รายรวมถึงกรณีที่รายงานตั้งแต่ปี 2020 5) มีการให้แนวทางการติดตามอาการทางปอด ตา และระบบประสาท
ประสิทธิภาพของการรักษาด้วยยาต้านการเกิดพังผืดด้วย nintedanib และ pirfenidone สำหรับพังผืดในปอดจาก HPS กำลังได้รับการประเมินอย่างต่อเนื่อง 4)
Flynn และคณะ (2025) รายงานว่าในการตรวจอวัยวะรับภาพของสตรีที่เป็นพาหะของการกลายพันธุ์ GP R143 ที่มี OA1 พบว่า 94% มีการเปลี่ยนแปลงของเม็ดสีแบบแผ่นก่อนแนวโค้ง และ 74% มีการส่องผ่านของม่านตา 3) ซึ่งแสดงให้เห็นว่าความผิดปกติทางตาเกิดขึ้นบ่อยแม้ในพาหะ
Q
ผู้ป่วยโรคเผือกมีความเสี่ยงสูงต่อการเป็นมะเร็งผิวหนังหรือไม่?
A
ใน OCA เนื่องจากการขาดเม็ดสีผิว ความเสียหายของดีเอ็นเอจากรังสียูวีจึงสะสมได้ง่าย ทำให้ความเสี่ยงของมะเร็งผิวหนัง เช่น มะเร็งเซลล์สความัส เพิ่มขึ้นอย่างมีนัยสำคัญ เชื่อกันว่าความเสี่ยงสูงเป็นพิเศษในผู้ป่วย OCA ที่มีเชื้อสายแอฟริกัน แนะนำให้พบแพทย์ผิวหนังเป็นประจำและป้องกันแสงแดดอย่างเคร่งครัด (ครีมกันแดด เสื้อผ้าปกปิด)
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCI D:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCI D:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCI D:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCI D:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCI D:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.