El albinismo es un grupo de trastornos hereditarios causados por mutaciones genéticas en la biosíntesis o el transporte de melanina, que resultan en una falta o reducción de pigmento en la piel, el cabello y los ojos. Además de la deficiencia de pigmento, este grupo de trastornos se caracteriza por anomalías oculares y de la vía visual específicas, como hipoplasia foveal, nistagmo y decusación anormal de la vía óptica.
No existe tratamiento curativo y la discapacidad visual es no progresiva (estática). En Japón, está designada como enfermedad intratable bajo la Ley de Enfermedades Intratables.
Prevalencia: Aproximadamente 1/17,000 en todo el mundo. En caucásicos, el AOC1 representa alrededor del 50%; en africanos, el AOC2 es el más común (1/10,000)1). En chinos han, el AOC1 es del 70.1% y el AOC2 del 10.2%1).
Albinismo ocular (AO)
Herencia recesiva ligada al cromosoma X: Afecta a varones. La pigmentación de la piel y el cabello es normal o solo ligeramente reducida, predominando los hallazgos oculares.
Mutación del gen GPR143: Implicado en la señalización de la biogénesis del melanosoma. Se han reportado 192 mutaciones3).
Mujeres portadoras: Muestran un patrón de hipopigmentación en mosaico similar a salpicaduras de barro en el fondo de ojo. Aproximadamente el 80% presenta anomalías del pigmento retiniano3).
Albinismo sindrómico
Síndrome de Hermansky-Pudlak (HPS): AOC + disfunción plaquetaria (tendencia al sangrado) + trastorno de la biogénesis de LRO. Existen 11 subtipos4, 5). Puede complicarse con fibrosis pulmonar y enfermedad inflamatoria intestinal.
Síndrome de Chediak-Higashi (CHS): AOC + inmunodeficiencia + trastornos neurológicos. Mutación del gen LYST.
12 genes: Se han identificado 12 genes para el albinismo sindrómico2).
La prevalencia mundial es de aproximadamente 1/17,0001), y en Europa se reporta alrededor de 1/12,0002). En japoneses, se conoce una distribución de AOC1 34%, AOC4 27% y HPS1 10%.
Q¿El albinismo es hereditario? ¿Puede ocurrir incluso si nadie en la familia tiene albinismo?
A
El OCA es un trastorno autosómico recesivo; si ambos padres son portadores con un alelo mutante cada uno, el hijo tiene un 25% de probabilidad de padecerlo. Dado que puede ocurrir incluso si ambos padres son fenotípicamente normales, no es infrecuente incluso sin antecedentes familiares. El OA es recesivo ligado al cromosoma X, afecta principalmente a varones, siendo la madre portadora. Las pruebas genéticas y el asesoramiento genético son importantes.
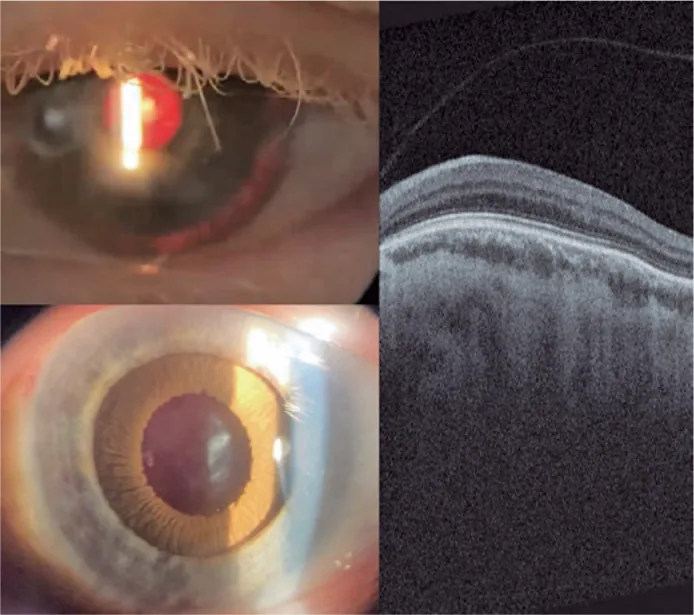
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Fotografía con lámpara de hendidura del ojo izquierdo 30 días después de la cirugía, que muestra el iris protésico sobre el surco ciliar mediante retroiluminación y después de midriasis farmacológica. OCT retiniana que muestra ausencia de fóvea y persistencia de las capas internas de la retina en el área esperada de la fóvea (hipoplasia macular).
Hipoplasia foveal: El factor más importante para la mala visión. La OCT confirma la ausencia de depresión foveal. En la angiografía fluoresceínica, la ausencia de zona avascular alrededor de la mácula también es un hallazgo característico. En OCA8, todos los casos muestran grado 3, y el grado se correlaciona con la agudeza visual2).
Transiluminación del iris: Debido a la falta de pigmento del iris, se observa aumento de la transiluminación en el examen con lámpara de hendidura. En OCA8, todos los casos muestran grado 32).
Nistagmo (INS): El nistagmo alternante periódico asimétrico se observa en aproximadamente el 10% de todos los INS, y hasta en el 37% en el INS del albinismo2).
Desvío anómalo del quiasma (chiasmal misrouting): Las fibras retinianas temporales que normalmente deberían seguir una vía ipsilateral (no cruzada) cruzan excesivamente al lado contralateral en el quiasma óptico. Se detecta mediante VEP. En OCA8, se han reportado coeficientes quiasmáticos de -0.97/-0.90, indicando asimetría severa2).
Estrabismo: Se observa en el 53-90.5% de los casos2).
Fondo de ojo hipopigmentado: Los vasos coroideos son visibles debido a la falta de pigmento en el epitelio pigmentario de la retina.
Fondo de ojo de portadores de OA1: El 94% muestra cambios pigmentarios parcheados anteriores a los vasos arcuatos, y el 74% muestra transiluminación del iris3). Pueden observarse manchas despigmentadas (fondo en mosaico) en la retina periférica, útiles para identificar portadores.
Q¿Cambia la agudeza visual en el albinismo a lo largo de la vida?
A
La disfunción visual debida al albinismo es estacionaria (no progresiva) y no empeora con la edad. Sin embargo, si no se realiza una corrección refractiva adecuada y un tratamiento para la ambliopía, la agudeza visual puede permanecer fija sin mejorar. La intervención temprana con corrección refractiva y cuidados de baja visión es importante.
Los principales genes causantes y las características de cada subtipo se muestran a continuación.
Subtipo
Gen causante
Características principales
OCA1
TYR
Deficiencia de tirosinasa
OCA2
OCA2 (gen P)
Regulación del pH del melanosoma1)
OCA3
TYRP1
Síntesis de eumelanina
OCA4
SLC45A2
Transportador de melanosomas
OCA8
DCT (TYRP2)
Conversión de dopacromo2)
OA1
GPR143
Señalización de melanosomas3)
HPS1/HPS4
HPS1/HPS4
Complejo BLOC-34)
HPS11
BLOC1S5
Complejo BLOC-15)
Función de la proteína OCA2: Pertenece a la familia de antiportadores Na⁺/H⁺ con estructura de 12 hélices α transmembrana 1). Como componente del canal aniónico específico del melanosoma, regula el pH de los melanosomas en estadio I/II mediante el control de la corriente de cloruro 1). ClinVar tiene registradas 477 mutaciones patogénicas 1).
DCT (TYRP2): Cataliza la conversión de dopacromo a DHICA (ácido 5,6-dihidroxiindol-2-carboxílico) 2). Es el gen causante del OCA8.
GPR143 (OA1): Molécula de señalización que regula el transporte vesicular de los melanosomas. Las mutaciones provocan la formación de macromelanosomas 3).
HPS (complejo BLOC): HPS1/HPS4 funcionan como un complejo BLOC-3 que actúa como GEF (factor de intercambio de nucleótidos de guanina) para Rab32/38 4). BLOC-1 consta de 8 subunidades y participa en el reciclaje endosomal; las mutaciones en el gen BLOC1S5 (BLOS3) causan HPS-11 (descrito por primera vez en 2020) 5).
Matrimonio consanguíneo: La prevalencia aumenta en poblaciones con alta tasa de consanguinidad, como en Pakistán 4).
Diferencias raciales/regionales: La frecuencia es alta en algunas partes de África.
Mutaciones heterocigotas compuestas: En OCA2 se han identificado nuevas mutaciones heterocigotas compuestas que contribuyen a la diversidad fenotípica 1).
Trialélico (OCA1): Combinaciones de tres alelos del gen TYR pueden dar lugar a OCA leve 2).
Q¿Por qué es importante conocer el tipo de albinismo?
A
Dado que el pronóstico visual, la presencia de complicaciones sistémicas y el patrón de herencia difieren según el tipo, el diagnóstico preciso del subtipo es importante. En particular, el HPS se asocia con tendencia al sangrado, fibrosis pulmonar y enfermedad inflamatoria intestinal, por lo que se requiere precaución antes de cirugía o extracción dental. La identificación del subtipo mediante pruebas genéticas conduce directamente a un plan de manejo adecuado.
El OCA típico se puede diagnosticar clínicamente por la combinación de cabello blanco, piel blanca y hallazgos oculares (transiluminación del iris, nistagmo). Sin embargo, en el OA1 japonés, el pigmento permanece en el iris, por lo que el diagnóstico puede retrasarse si los pacientes se presentan solo con síntomas oculares.
VEP (Potencial Evocado Visual): Más importante para detectar la decusación anormal de la vía óptica. Se utiliza VEP de 3 derivaciones para evaluar la asimetría de la forma de onda entre los hemisferios izquierdo y derecho 2). Se cuantifica mediante el coeficiente quiasmático (−1 indica máxima asimetría, +1 normal) 2).
Medición del ángulo lambda: Un ángulo lambda >5 grados es un indicador clínico fuerte de albinismo 2).
Microscopía electrónica de plaquetas: Se utiliza para el diagnóstico definitivo de HPS. La ausencia de cuerpos densos es un hallazgo característico 4).
Pruebas genéticas: Secuenciación del exoma completo (WES) 1) o secuenciación de panel 2, 4, 5). Particularmente importante cuando el fenotipo es leve o atípico.
No existe un tratamiento curativo. El objetivo del tratamiento es maximizar la función visual y manejar las complicaciones.
Manejo Visual
Corrección refractiva: La base es la corrección con gafas desde la infancia. La intervención temprana para prevenir la ambliopía es importante.
Gafas tintadas: Use lentes que bloqueen aproximadamente el 20% de la luz en interiores y aproximadamente el 80% en exteriores 5).
Lentes de contacto con iris: Se pueden usar para mejorar la apariencia y reducir la fotofobia.
Cuidado de baja visión: Utilice dispositivos de ayuda como lupas, magnificadores de lectura y tabletas.
Nistagmo y Estrabismo
Cirugía de estrabismo: Se realiza con fines cosméticos. En algunos casos, se puede realizar la cirugía de Dawson-Trick-Litzkow (DTL) para reducir el nistagmo.
Tratamiento del nistagmo: No existe un tratamiento curativo. Se puede considerar la cirugía de estrabismo para abordar la postura anormal de la cabeza (zona nula).
Manejo Cutáneo y Sistémico
Protección solar: Protección de la piel con protector solar que bloquee los rayos UVB, ropa y sombreros. Incluso en personas japonesas, se debe prestar atención al riesgo de carcinoma de células escamosas.
Manejo del HPS: Los AINE (incluyendo la aspirina) están generalmente contraindicados porque empeoran la disfunción plaquetaria. Se recomienda la monitorización regular de la función pulmonar desde la adolescencia 4).
Consejo genético: Esencial para la planificación familiar, el diagnóstico de portadores y la determinación del subtipo.
Se utilizan agentes antifibróticos para la fibrosis pulmonar asociada con HPS.
Liu et al. (2025) informaron que nintedanib estabilizó la fibrosis pulmonar durante 18 meses en un caso de HPS con una nueva mutación homocigótica de HPS4 4). La pirfenidona también es una opción de tratamiento utilizada de manera similar.
Q¿Está disponible clínicamente la terapia génica para el albinismo?
A
Actualmente, la terapia génica para el albinismo no está disponible clínicamente. La terapia génica ha sido aprobada para otras enfermedades retinianas hereditarias como la retinosquisis y la LCA (ej. Luxturna), y se están realizando investigaciones sobre su aplicación al albinismo 6). Actualmente, la corrección refractiva y el cuidado de baja visión son el centro del tratamiento estándar.
La síntesis de melanina ocurre dentro de los melanosomas a través de la siguiente vía:
Tirosina → L-DOPA → DOPA quinona → (eumelanina o feomelanina)
La conversión de dopacromo a DHICA (ácido 5,6-dihidroxiindol-2-carboxílico) es catalizada por DCT (TYRP2) 2), una reacción esencial para la síntesis de eumelanina.
Defecto de maduración del melanosoma en OCA2: La función reducida de la proteína OCA2 altera la transición de los melanosomas a la etapa IV (melanosomas maduros), aumentando el número de melanosomas inmaduros en etapa I/II 1). La alteración de la regulación del pH mediante el control de la corriente de cloruro es el mecanismo principal 1).
Cuando la pigmentación del epitelio pigmentario de la retina es inadecuada, se pierde el efecto de protección contra la luz durante el desarrollo, lo que dificulta el desarrollo morfológico de la fóvea (formación de la zona avascular y empaquetamiento denso de los conos). Esta es la causa fundamental de la mala visión en el albinismo.
Mecanismo de la Decusación Anormal de la Vía Óptica
En el albinismo, las fibras de la retina temporal cruzan excesivamente al lado contralateral en el quiasma óptico, de modo que más fibras de lo normal se procesan como fibras nasales. Este ‘misrouting’ se detecta mediante VEP como asimetría del coeficiente quiasmático 2).
En su estudio de OCA8 de 2025, Rateaux et al. utilizaron un modelo de ratón en el que la mutación de DCT reduce la L-DOPA al 50% de los niveles de tipo salvaje y demostraron que la suplementación con L-DOPA restaura las anomalías oftálmicas 2). Esto sugiere un papel de la L-DOPA como intermediario de la síntesis de melanina en la formación de la vía óptica.
Mutación de GPR143 (OA1): La pérdida de función de la proteína GPR143 altera el transporte vesicular del melanosoma, lo que lleva a la formación de macromelanosomas 3).
HPS (trastornos del complejo BLOC): La pérdida de función de BLOC-3 altera la activación de Rab32/38, lo que resulta en una interrupción general de la biogénesis de los orgánulos relacionados con el melanosoma (LRO: orgánulos relacionados con lisosomas) 4). Los cuerpos densos en las plaquetas y los cuerpos lamelares en las células epiteliales pulmonares tipo II se ven afectados. Se cree que la acumulación de sustancias similares a ceroides a partir de cuerpos lamelares anormales está involucrada en el contexto de la fibrosis pulmonar 4).
BLOC-1 (HPS-11): Funciona como un complejo de ocho subunidades que incluye BLOC1S5 (BLOS3) y regula el reciclaje endosomal 5).
7. Investigación más reciente y perspectivas futuras (etapa de investigación)
La L-DOPA es un intermediario en la síntesis de melanina y también desempeña un papel importante en el desarrollo de la retina y la formación de la vía óptica.
En el modelo de ratón OCA8 de Rateaux et al. (2025), la reducción de L-DOPA debida a la mutación de DCT fue la causa principal del fenotipo ocular, y la suplementación con L-DOPA mejoró las anomalías 2). Se está estudiando su aplicación en humanos 6).
Se está llevando a cabo una evaluación de la mejora de la función visual mediante la administración de levodopa en pacientes con albinismo, y se espera un efecto promotor del desarrollo de la retina6).
La nitisinona, un inhibidor de la 4-hidroxifenilpiruvato dioxigenasa (4-HPPD), se está estudiando por su potencial para aumentar la melanina ocular al modular la vía del metabolismo de la tirosina 6).
La investigación básica sobre la terapia de reemplazo génico dirigida a genes relacionados con OCA (TYR, OCA2, etc.) está avanzando. La aprobación de Luxturna (reemplazo del gen RPE65) para LCA (amaurosis congénita de Leber) está acelerando la investigación de terapia génica para enfermedades hereditarias de la retina en general 6).
Jiang et al. (2024) identificaron una nueva mutación compuesta heterocigota (c.635A>G/c.2359+1G>T) en el gen OCA2 mediante secuenciación del exoma completo (WES) en una familia china 1). El análisis funcional confirmó que la proteína mutante tiene un transporte alterado a los melanocitos en comparación con la de tipo salvaje.
Descripción e investigación terapéutica del HPS-11
Boeckelmann et al. (2021) detallaron el espectro clínico del HPS-11 causado por mutaciones en BLOC1S5 en un estudio de 5 casos, incluidos los reportados después de 2020 5). Se han proporcionado pautas para el monitoreo de los síntomas pulmonares, oculares y neurológicos.
La eficacia de la terapia antifibrótica con nintedanib y pirfenidona para la fibrosis pulmonar en HPS se evalúa continuamente 4).
Flynn et al. (2025) informaron que en los exámenes de fondo de ojo de mujeres portadoras de OA1 con mutaciones en GPR143, el 94% presentaba cambios pigmentarios parcheados arqueados prepapilares y el 74% presentaba transiluminación del iris3). Se demostró que las anomalías oculares son frecuentes incluso en portadores.
Q¿Los pacientes con albinismo tienen un alto riesgo de cáncer de piel?
A
En OCA, debido a la falta de pigmento cutáneo, el daño del ADN por radiación ultravioleta se acumula fácilmente, aumentando significativamente el riesgo de cánceres de piel como el carcinoma de células escamosas. Se considera que el riesgo es particularmente alto en pacientes africanos con OCA. Se recomiendan visitas regulares al dermatólogo y una protección solar exhaustiva (protector solar, ropa protectora).
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.