백색증(Albinism)은 멜라닌 색소의 생합성 또는 수송에 관여하는 유전자 변이로 인해 피부, 모발, 눈의 색소가 결핍 또는 감소하는 유전성 질환군입니다. 색소 결핍에 더하여 중심와 형성부전, 안진, 시각로 이상 교차 등의 특유한 안구 및 시각로 이상을 동반하는 점이 이 질환군의 특징입니다.
근치적 치료는 없으며, 시기능 장애는 비진행성(정지성)입니다. 일본에서는 지정 난치병(난치병법)으로 지정되어 있습니다.
GPR143 유전자 돌연변이: 멜라노솜 생합성 신호 전달에 관여합니다. 192개의 돌연변이가 보고되었습니다3).
보인자 여성: 안저에 진흙 튄 모자이크 모양의 저색소 패턴을 보입니다. 약 80%에서 망막 색소 이상이 관찰됩니다3).
증후군성 백색증
Hermansky-Pudlak 증후군(HPS): OCA + 혈소판 기능 이상(출혈 경향) + LRO 생합성 장애. 11개의 아형이 있습니다4, 5). 폐섬유증 및 염증성 장 질환이 동반될 수 있습니다.
Chediak-Higashi 증후군(CHS): OCA + 면역 결핍 + 신경 장애. LYST 유전자 돌연변이.
12개 유전자: 증후군성 백색증에 대해 12개의 유전자가 확인되었습니다2).
전 세계 유병률은 약 1/17,000이며1), 유럽에서는 약 1/12,000으로 보고됩니다2). 일본인에서는 OCA1 34%, OCA4 27%, HPS1 10%의 분포가 알려져 있습니다.
Q백색증은 유전되나요? 가족 중에 백색증 환자가 없어도 발병할 수 있나요?
A
OCA는 상염색체 열성 유전으로, 양쪽 부모가 각각 하나의 돌연변이 대립유전자를 보유한 보인자인 경우 자녀에게 25%의 확률로 발병합니다. 부모의 표현형이 정상이더라도 발병할 수 있으므로 가족력이 없어도 드물지 않습니다. OA는 X-연관 열성 유전으로 주로 남성에게 발병하며, 어머니가 보인자입니다. 유전자 검사와 유전 상담이 중요합니다.
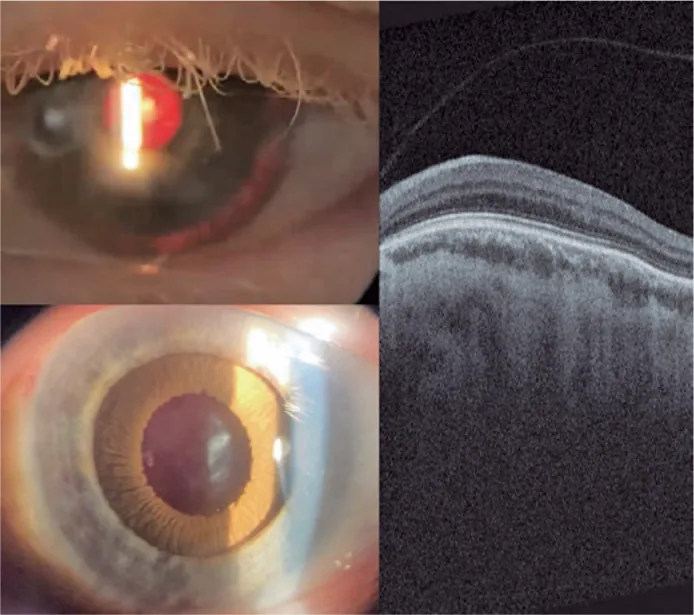
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
수술 후 30일째 좌안 세극등 사진, 후방 조명 및 약물 산동 후 섬모체 고랑에 있는 인공 홍채를 보여줍니다. 망막OCT는 중심와 함몰의 부재와 예상 중심와 영역을 통한 망막 내층의 지속(황반 저형성)을 보여줍니다.
중심와 형성부전: 시력 저하의 가장 중요한 요인입니다. OCT에서 중심와 함몰의 결여를 확인합니다. 형광 안저 촬영에서는 황반 주변 무혈관 영역이 보이지 않는 것도 특징적인 소견입니다. OCA8에서는 모든 예에서 3등급을 보이며, 등급과 시력은 상관관계가 있습니다2).
홍채 투과: 홍채 색소 결여로 인해 세극등 현미경에서 투과성 항진이 관찰됩니다. OCA8에서는 모든 예에서 3등급입니다2).
안진 (INS): 비대칭 주기 교대 안진은 전체 INS의 약 10%에서 나타나며, 백색증 INS에서는 최대 37%에 이릅니다2).
시각 경로 이상 교차 (chiasmal misrouting): 본래 동측(교차되지 않은) 경로를 따라야 할 측두 망막 섬유가 시교차에서 반대측으로 과도하게 교차합니다. VEP로 검출합니다. OCA8에서는 시교차 계수가 -0.97/-0.90으로 심한 비대칭이 보고되었습니다2).
사시: 53-90.5%에서 관찰됩니다2).
저색소 안저: 망막 색소 상피의 색소 결여로 인해 맥락막 혈관이 투시됩니다.
OA1 보인자의 안저: 94%에서 궁상 혈관 전방의 반점상 색소 변화, 74%에서 홍채 투과가 관찰됩니다3). 주변부 망막에 탈색소 반점(모자이크 안저)이 나타날 수 있으며, 보인자 식별에 유용합니다.
Q백색증의 시력은 평생 동안 변화합니까?
A
백색증으로 인한 시각 기능 장애는 정지성(비진행성)이며 나이가 들어도 악화되지 않습니다. 그러나 적절한 굴절 교정이나 약시 치료가 이루어지지 않으면 시력이 개선되지 않고 고정될 수 있습니다. 굴절 교정 및 저시력 관리의 조기 개입이 중요합니다.
복합 이형접합 돌연변이: OCA2에서 새로운 복합 이형접합 돌연변이가 확인되었으며, 표현형 다양성에 관여함1).
3대립유전자형 (OCA1): TYR 유전자의 3개 대립유전자 조합으로 경증 OCA가 발생할 수 있음2).
Q백색증의 유형을 아는 것이 왜 의미가 있나요?
A
유형에 따라 시력 예후, 전신 합병증 유무, 유전 양식이 다르므로 정확한 아형 진단이 중요합니다. 특히 HPS는 출혈 경향, 폐섬유증, 염증성 장 질환을 동반하므로 수술이나 발치 전 주의가 필요합니다. 유전자 검사를 통한 아형 확인은 적절한 관리 계획 수립에 직접 연결됩니다.
Liu 등(2025)은 새로운 동형접합 HPS4 돌연변이를 가진 HPS 증례에서 닌테다닙 투여로 폐섬유증이 18개월간 안정화되었음을 보고했습니다4). 피르페니돈도 유사하게 사용되는 치료 선택지입니다.
Q백색증에 대한 유전자 치료가 실용화되었습니까?
A
현재 백색증에 대한 유전자 치료는 실용화되지 않았습니다. 망막분리증이나 LCA와 같은 다른 유전성 망막 질환에서는 유전자 치료가 승인되었으며(예: Luxturna), 백색증에 대한 응용 연구가 진행 중입니다6). 현재는 굴절 교정과 저시력 관리가 표준 치료의 중심입니다.
백색증에서는 관자쪽 망막에서 나온 섬유가 시교차에서 과도하게 반대쪽으로 교차하여, 평소보다 더 많은 섬유가 코쪽 섬유로 처리됩니다. 이 ‘misrouting’은 VEP에서 시교차 계수의 비대칭으로 포착됩니다2).
Rateaux 등(2025)의 OCA8 연구에서는 DCT 돌연변이로 인해 L-DOPA가 야생형의 50%로 감소하는 마우스 모델을 사용하여 L-DOPA 보충이 안과적 이상을 회복시킴을 보여주었습니다2). L-DOPA가 멜라닌 합성 중간체로서 시각 경로 형성에 역할을 한다는 것이 시사됩니다.
GPR143 돌연변이(OA1) : GPR143 단백질의 기능 상실로 인해 멜라닌소체 소포 수송이 손상되어 거대멜라닌소체가 형성됩니다3).
HPS(BLOC 복합체 장애) : BLOC-3의 기능 상실로 Rab32/38의 활성화가 손상되어 멜라닌소체 관련 세포소기관(LRO: 리소좀 관련 세포소기관)의 생합성이 전반적으로 붕괴됩니다4). 혈소판 내 치밀소체와 폐 II형 상피세포 내 라멜라소체가 영향을 받습니다. 폐섬유증의 배경에는 비정상적인 라멜라소체로부터의 세로이드 유사 물질 축적이 관여하는 것으로 알려져 있습니다4).
BLOC-1(HPS-11) : BLOC1S5(BLOS3)를 포함한 8개 서브유닛 복합체로 기능하며 엔도솜 재활용을 조절합니다5).
Jiang 등(2024)은 중국인 가족의 전장 엑솜 시퀀싱(WES)을 통해 OCA2 유전자의 새로운 복합 이형접합 변이(c.635A>G/c.2359+1G>T)를 동정했습니다1). 기능 분석 결과, 변이 단백질은 야생형에 비해 멜라닌세포로의 이동이 손상되는 것으로 확인되었습니다.
Flynn 등(2025)은 GPR143 변이를 가진 여성 OA1 보인자의 안저 검사에서 94%에서 궁상 시신경유두전 반점상 색소 변화, 74%에서 홍채 투과조명이 확인되었다고 보고했습니다3). 보인자에서도 안과적 이상이 높은 비율로 관찰되는 것으로 나타났습니다.
Q백색증 환자는 피부암 위험이 높습니까?
A
OCA에서는 피부 색소가 결여되어 자외선에 의한 DNA 손상이 축적되기 쉬우며, 편평세포암 등의 피부암 위험이 유의하게 증가합니다. 아프리카계 OCA 환자에서 특히 위험이 높은 것으로 알려져 있습니다. 정기적인 피부과 진료와 철저한 자외선 차단(자외선 차단제, 차폐 의복)이 권장됩니다.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.