Albinizm, melanin pigmentinin biyosentezi veya taşınmasında rol oynayan genlerdeki mutasyonlar nedeniyle cilt, saç ve gözlerde pigmentin eksik veya azalmış olduğu kalıtsal bir hastalık grubudur. Pigment eksikliğine ek olarak, foveal hipoplazi, nistagmus ve anormal optik yolak çaprazlanması gibi karakteristik göz ve görme yolu anormallikleri bu hastalık grubunun özelliğidir.
Kesin tedavisi yoktur ve görme bozukluğu ilerleyici değildir (durağandır). Japonya’da belirlenmiş nadir hastalık (Nadir Hastalıklar Yasası) kapsamındadır.
Otozomal resesif kalıtım: En sık görülen grup. Deri, saç ve gözlerde pigment eksikliğine neden olur.
8 gen ve 8 alt tip: OKA1 (TYR), OKA2 (OKA2/P geni), OKA3 (TYRP1), OKA4 (SLC45A2), OKA8 (DCT) vb.
Prevalans: Dünyada yaklaşık 1/17.000. Beyazlarda OKA1 yaklaşık %50’sini oluştururken, Afrika kökenlilerde OKA2 en sıktır (1/10.000)1). Çin Hanlarında OKA1 %70,1, OKA2 %10,2’dir1).
Oküler Albinizm (OA)
X’e bağlı resesif kalıtım: Erkeklerde görülür. Deri ve saç pigmenti normal veya hafif azalmıştır, göz bulguları baskındır.
GPR143 gen mutasyonu: Melanozom biyosentez sinyal iletiminde rol oynar. 192 mutasyon rapor edilmiştir3).
Taşıyıcı kadınlar: Fundusta çamur sıçramışı benzeri mozaik hipopigmentasyon paterni gösterir. Yaklaşık %80’inde retina pigment anormalliği bulunur3).
Sendromik Albinizm
Hermansky-Pudlak sendromu (HPS): OKA + trombosit fonksiyon bozukluğu (kanama eğilimi) + LRO biyosentez bozukluğu. 11 alt tipi vardır4, 5). Pulmoner fibrozis ve inflamatuar bağırsak hastalığı eşlik edebilir.
Chediak-Higashi sendromu (CHS): OKA + immün yetmezlik + nörolojik bozukluk. LYST gen mutasyonu.
12 gen: Sendromik albinizm için 12 gen tanımlanmıştır2).
Dünya prevalansı yaklaşık 1/17.000’dir1) ve Avrupa’da yaklaşık 1/12.000 olarak bildirilmiştir2). Japonlarda OKA1 %34, OKA4 %27 ve HPS1 %10 dağılımı bilinmektedir.
QAlbinizm kalıtsal mıdır? Ailede albinizmli biri olmasa bile ortaya çıkabilir mi?
A
OKA, otozomal resesif kalıtımlıdır ve her iki ebeveyn de birer mutant alel taşıyıcısı ise, çocukta %25 olasılıkla hastalık görülür. Ebeveynler fenotipik olarak normal olsa bile hastalık ortaya çıkabileceğinden, aile öyküsü olmaması nadir değildir. OA, X’e bağlı resesif kalıtımlıdır ve esas olarak erkeklerde görülür, anne taşıyıcıdır. Genetik test ve genetik danışmanlık önemlidir.
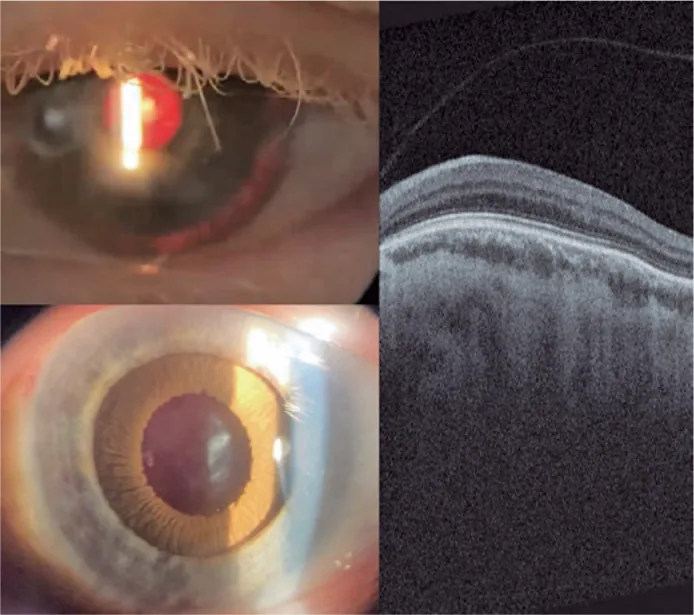
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Ameliyattan 30 gün sonra sol göz yarık lamba fotoğrafı, retrolüminasyon ve farmakolojik midriyazis sonrası siliyer sulkusta protez iris görülüyor. RetinaOCT’si, foveal çukurun olmadığını ve beklenen fovea alanında retina iç katmanlarının devam ettiğini (maküler hipoplazi) göstermektedir.
Foveal hipoplazi: Kötü görmenin en önemli faktörü. OCT’de foveal çukurluğun olmadığı görülür. Floresein anjiyografide makula çevresinde avasküler alanın olmaması da karakteristik bir bulgudur. OCA8’de tüm vakalar grade 3’tür ve grade ile görme koreledir 2).
İris transilluminasyonu: İris pigmentinin olmaması nedeniyle yarık lamba biyomikroskopisinde transilluminasyon artışı görülür. OCA8’de tüm vakalar grade 3’tür 2).
Nistagmus (INS): Asimetrik periyodik alternan nistagmus tüm INS vakalarının yaklaşık %10’unda, albinizm INS’de ise %37’ye kadar görülür 2).
Kiazmal yanlış yönlenme (chiasmal misrouting): Normalde ipsilateral (çaprazlaşmamış) yolu izlemesi gereken temporal retina lifleri, optik kiazmada aşırı derecede karşı tarafa çaprazlaşır. VEP ile tespit edilir. OCA8’de kiazmal indeks -0.97/-0.90 ile ileri derecede asimetri bildirilmiştir 2).
Şaşılık: %53-90.5 oranında görülür 2).
Hipopigmente fundus: Retina pigment epitelindeki pigment eksikliği nedeniyle koroid damarları görülebilir.
Albminizme bağlı görme bozukluğu statiktir (ilerleyici değildir) ve yaşla birlikte kötüleşmez. Ancak uygun refraksiyon düzeltmesi veya ambliyopi tedavisi yapılmazsa görme düzelmeden sabit kalabilir. Erken dönemde refraksiyon düzeltmesi ve az görme rehabilitasyonu önemlidir.
GPR143 (OA1): Melanozom vezikül taşınmasını düzenleyen bir sinyal molekülüdür. Mutasyonu makromelanozom oluşumuna yol açar 3).
HPS (BLOC kompleksi): HPS1/HPS4, BLOC-3 kompleksi olarak Rab32/38 için GEF (guanin nükleotid değişim faktörü) işlevi görür 4). BLOC-1, 8 alt birimden oluşur ve endozom geri dönüşümünde rol alır; BLOC1S5 (BLOS3) gen mutasyonu HPS-11’e (ilk kez 2020’de tanımlanmıştır) neden olur 5).
Akraba evliliği: Pakistan gibi akraba evliliğinin yaygın olduğu toplumlarda prevalans artar 4).
Irk ve bölgesel farklılıklar: Afrika’nın bazı bölgelerinde sıklık yüksektir.
Bileşik heterozigot mutasyonlar: OCA2’de yeni bileşik heterozigot mutasyonlar tanımlanmış olup fenotipik çeşitlilikle ilişkilidir 1).
Üç alelli tip (OCA1): TYR genindeki üç alel kombinasyonu hafif OCA’ya neden olabilir 2).
QAlbinizm tipini bilmek neden önemlidir?
A
Görme prognozu, sistemik komplikasyonların varlığı ve kalıtım şekli tipe göre farklılık gösterdiğinden, doğru alt tip tanısı önemlidir. Özellikle HPS’de kanama eğilimi, pulmoner fibrozis ve inflamatuar bağırsak hastalığı görülür; cerrahi veya diş çekimi öncesinde dikkatli olunmalıdır. Genetik test ile alt tipin belirlenmesi, uygun yönetim planının oluşturulmasına doğrudan katkı sağlar.
Tipik OCA, beyaz saç, beyaz cilt ve göz bulguları (iris transilluminasyonu, nistagmus) kombinasyonu ile klinik olarak teşhis edilebilir. Ancak Japon OA1’de iriste pigment kalır, bu nedenle sadece göz semptomlarıyla başvuran hastalarda tanı gecikebilir.
Yarık lamba biyomikroskobu : İris transilluminasyonunun doğrulanması. Derecelendirme yapılır.
OCT (Optik Koherens Tomografi) : Foveal hipoplazinin derecelendirilmesi için gereklidir. Çocuklarda da uygulanabilir ve erken müdahale kararına yardımcı olur 6).
VEP (Görsel Uyarılmış Potansiyel) : Anormal optik yolak çaprazlanmasının saptanmasında en önemli yöntemdir. Üç kanallı VEP ile sol ve sağ hemisfer dalga formu asimetrisi değerlendirilir 2). Kiazma katsayısı (‒1 maksimum asimetri, +1 normal) ile kantifiye edilir 2).
Lambda açısı ölçümü : Lambda açısının 5 dereceden büyük olması albinizmi güçlü bir şekilde düşündüren klinik bir göstergedir 2).
Kesin bir tedavi yoktur. Tedavinin amacı görme fonksiyonunu en üst düzeye çıkarmak ve komplikasyonları yönetmektir.
Görme Yönetimi
Kırma kusuru düzeltilmesi: Bebeklik ve çocukluk döneminden itibaren gözlükle düzeltme temeldir. Ambliyopiyi önlemek için erken müdahale önemlidir.
Işık koruyucu gözlükler: İç mekanda yaklaşık %20, dış mekanda yaklaşık %80 ışık geçirgenliğini azaltan lensler kullanılır5).
Renkli kontakt lensler: Kozmetik iyileşme ve fotofobiyi azaltmak amacıyla kullanılabilir.
Az görme rehabilitasyonu: Büyüteç, büyüteçli okuma cihazı, tablet gibi yardımcı araçlar kullanılır.
Nistagmus ve Şaşılık
Şaşılık cerrahisi: Kozmetik amaçlı yapılır. Nistagmusu azaltmak için Dawson-Trick-Litzkow (DTL) ameliyatı da uygulanabilir.
Nistagmus tedavisi: Kesin tedavisi yoktur. Anormal baş pozisyonu (null zone sağlanması) için şaşılık cerrahisi düşünülür.
Deri ve Sistemik Yönetim
Güneşten korunma: UVB bloke edici güneş kremi, giysi ve şapka ile cilt koruması. Japonlarda da skuamöz hücreli karsinom riskine dikkat edilmelidir.
HPS yönetimi: NSAİİ’ler (aspirin dahil) trombosit fonksiyon bozukluğunu kötüleştirdiği için genellikle kontrendikedir. Ergenlikten itibaren düzenli akciğer fonksiyon takibi önerilir4).
Genetik danışmanlık: Aile planlaması, taşıyıcı tanısı ve alt tip belirleme için gereklidir.
HPS’ye eşlik eden akciğer fibrozisinde antifibrotik ilaçlar kullanılır.
Liu ve ark. (2025), yeni bir homozigot HPS4 mutasyonu taşıyan HPS olgusunda nintedanib uygulamasıyla akciğer fibrozisinin 18 ay boyunca stabilize olduğunu bildirmiştir4). Pirfenidon da benzer şekilde kullanılan bir tedavi seçeneğidir.
QAlbinizm için gen tedavisi uygulamaya konuldu mu?
A
Şu anda albinizm için gen tedavisi uygulamaya konulmamıştır. Retinoskizis ve LCA gibi diğer kalıtsal retina hastalıklarında gen tedavisi onaylanmıştır (örneğin Luxturna) ve albinizme uygulanması için araştırmalar devam etmektedir6). Şu anda refraktif düzeltme ve az görme rehabilitasyonu standart tedavinin merkezindedir.
Melanin sentezi, melanozomlar içinde aşağıdaki yolu izler:
Tirozin → L-DOPA → DOPA kinon → (Ömelanin veya Feomelanin)
Dopakromdan DHICA’ya (5,6-dihidroksiindol-2-karboksilik asit) dönüşüm, DCT (TYRP2) tarafından katalize edilir2) ve ömelanin sentezi için gerekli bir reaksiyondur.
OCA2’de Melanozom Olgunlaşma Bozukluğu: OCA2 proteininin işlevindeki azalma, melanozomların evre IV (olgun melanozom) geçişini bozar ve evre I/II olgunlaşmamış melanozomların artmasına neden olur1). Klor akımı kontrolü yoluyla pH düzenlemesinin bozulması ana mekanizmadır1).
Retina pigment epitelinin pigmentasyonu uygunsuz olduğunda, gelişim sırasında ışık koruyucu etki kaybolur ve foveanın morfolojik gelişimi (avasküler zon oluşumu ve koni hücre yoğunlaşması) engellenir. Bu, albinizmdeki zayıf görmenin temel nedenidir.
Albinizmde, temporal retinadan gelen lifler optik kiazmada aşırı derecede karşı tarafa çaprazlaşır ve normalden daha fazla lif nazal lif olarak işlenir. Bu ‘yanlış yönlendirme’, VEP’de kiazmatik katsayının asimetrisi olarak tespit edilebilir2).
Rateaux ve ark. (2025) tarafından yapılan OCA8 çalışmasında, DCT mutasyonu nedeniyle L-DOPA seviyesi vahşi tipin %50’sine düşen fare modeli kullanılarak, L-DOPA takviyesinin oftalmik anormallikleri düzelttiği gösterilmiştir 2). L-DOPA’nın melanin sentez ara ürünü olarak görme yolu oluşumunda oynadığı rol öne sürülmektedir.
HPS (BLOC kompleks bozukluğu): BLOC-3 işlev kaybı, Rab32/38 aktivasyonunu bozarak melanozomla ilişkili organellerin (LRO: lizozomla ilişkili organel) biyosentezinde genel bir bozulmaya neden olur 4). Trombositlerdeki yoğun cisimcikler ve akciğer tip II epitel hücrelerindeki lameller cisimcikler etkilenir. Akciğer fibrozunun arka planında, anormal lameller cisimciklerden seroid benzeri madde birikiminin rol oynadığı düşünülmektedir 4).
BLOC-1 (HPS-11): BLOC1S5 (BLOS3) dahil 8 alt birimli bir kompleks olarak işlev görür ve endozomal geri dönüşümü düzenler 5).
7. Güncel Araştırmalar ve Gelecek Perspektifleri (Araştırma Aşamasındaki Raporlar)
L-DOPA, melanin sentezinde bir ara ürün olmasının yanı sıra retina gelişimi ve görme yolu oluşumunda önemli bir rol oynar.
Rateaux ve ark. (2025) tarafından yapılan OCA8 fare modelinde, DCT mutasyonuna bağlı L-DOPA düşüklüğü oftalmik fenotipin ana nedeniydi ve L-DOPA takviyesi anormallikleri düzeltti 2). İnsanlarda uygulanması araştırılmaktadır 6).
Albinizm hastalarında levodopa uygulaması ile görme işlevinde iyileşme değerlendirmesi devam etmekte olup, retina gelişimini hızlandırıcı etkiler beklenmektedir 6).
4-hidroksifenilpiruvat dioksijenaz (4-HPPD) inhibitörü olan nitisinon, tirozin metabolizma yolunu düzenleyerek göz içi melanini artırma potansiyeli açısından araştırılmaktadır 6).
OCA ile ilişkili genleri (TYR, OCA2 vb.) hedef alan gen replasman tedavisine yönelik temel araştırmalar ilerlemektedir. LCA (Leber konjenital amorozu) için Luxturna’nın (RPE65 gen replasmanı) onayı, kalıtsal retina hastalıklarının gen tedavisi araştırmalarını genel olarak hızlandırmıştır 6).
Jiang ve ark. (2024), Çinli bir ailede tüm ekzom dizileme (WES) ile OCA2 geninde yeni bir bileşik heterozigot mutasyon (c.635A>G/c.2359+1G>T) tanımlamıştır 1). Fonksiyonel analiz, mutant proteinin vahşi tipe kıyasla melanositlere translokasyonunun bozulduğunu doğrulamıştır.
Boeckelmann ve ark. (2021), BLOC1S5 mutasyonuna bağlı HPS-11’in klinik spektrumunu, 2020’den sonra bildirilen vakaları da içeren 5 vakanın incelenmesiyle ayrıntılı olarak tanımlamıştır 5). Akciğer, göz ve nörolojik semptomların izlenmesi için kılavuzlar sağlanmıştır.
HPS pulmoner fibrozisi için nintedanib ve pirfenidon ile antifibrotik tedavinin etkinliği sürekli olarak değerlendirilmektedir 4).
Flynn ve ark. (2025), GPR143 mutasyonu olan OA1 taşıyıcısı kadınların fundus muayenesinde %94’ünde arkuat ön benekli pigment değişiklikleri ve %74’ünde iris transilluminasyonu bildirmiştir 3). Taşıyıcılarda bile oküler anormalliklerin yüksek oranda görüldüğü gösterilmiştir.
QAlbinizmli hastalar cilt kanseri riski altında mıdır?
A
OCA’da cilt pigmenti eksikliği nedeniyle UV kaynaklı DNA hasarı kolayca birikir ve skuamöz hücreli karsinom gibi cilt kanseri riski önemli ölçüde artar. Afrika kökenli OCA hastalarında risk özellikle yüksektir. Düzenli dermatolojik muayene ve kapsamlı güneş koruması (güneş kremi, koruyucu giysi) önerilir.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.