眼皮膚白化症(OCA)
體染色體隱性遺傳:最常見的一群。皮膚、毛髮和眼睛均出現色素缺乏。
8個基因、8種亞型:OCA1(TYR)、OCA2(OCA2/P基因)、OCA3(TYRP1)、OCA4(SLC45A2)、OCA8(DCT)等。
盛行率:全球約1/17,000。白人中OCA1約佔50%,非洲裔中OCA2最常見(1/10,000)1)。中國漢族中OCA1佔70.1%,OCA2佔10.2%1)。

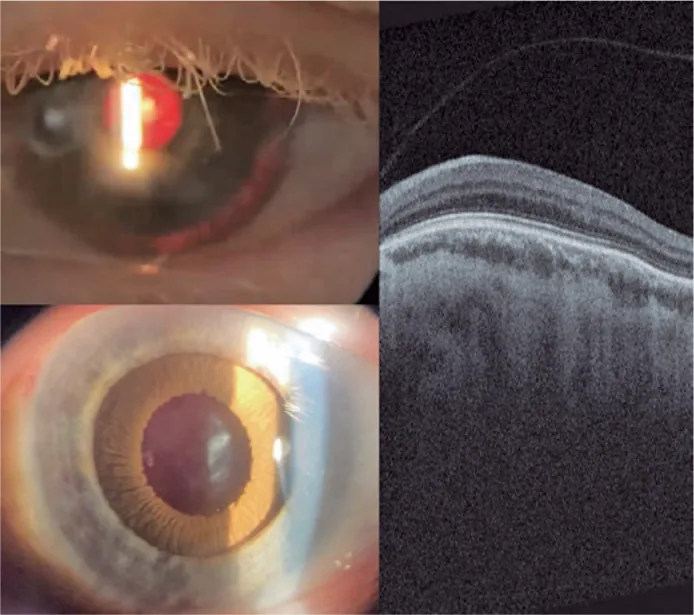
白化症(Albinism)是由於黑色素生合成或運輸相關基因突變,導致皮膚、毛髮、眼睛色素缺乏或減少的一組遺傳性疾病。除色素缺乏外,本組疾病的特點還包括中心凹發育不全、眼球震顫及視路異常交叉等特有的眼與視路異常。
無根治性治療,視功能障礙為非進行性(靜止性)。在日本被指定為指定難病(難病法)。
白化症大致分為三類。
眼皮膚白化症(OCA)
體染色體隱性遺傳:最常見的一群。皮膚、毛髮和眼睛均出現色素缺乏。
8個基因、8種亞型:OCA1(TYR)、OCA2(OCA2/P基因)、OCA3(TYRP1)、OCA4(SLC45A2)、OCA8(DCT)等。
盛行率:全球約1/17,000。白人中OCA1約佔50%,非洲裔中OCA2最常見(1/10,000)1)。中國漢族中OCA1佔70.1%,OCA2佔10.2%1)。
眼白化症(OA)
X染色體隱性遺傳:男性發病。皮膚和毛髮色素正常或僅輕度減少,以眼部表現為主。
GPR143基因突變:參與黑素體生物合成訊號傳導。已報告192種突變3)。
女性攜帶者:眼底呈現泥濺樣鑲嵌狀低色素模式。約80%有視網膜色素異常3)。
症候群性白化症
全球盛行率約為1/17,0001),歐洲報告約為1/12,0002)。日本人中分布已知為OCA1佔34%、OCA4佔27%、HPS1佔10%。
OCA為體染色體隱性遺傳,若父母雙方皆為帶有一個突變等位基因的帶因者,子女有25%的機率發病。即使父母表現型正常也可能發病,因此無家族史的情況並不少見。OA為X染色體隱性遺傳,主要影響男性,母親為帶因者。基因檢測與遺傳諮詢非常重要。
各主要亞型之視力參考範圍如下所示。
| 亞型 | 視力範圍參考 |
|---|---|
| OCA1 | 多為0.1以下 |
| OCA2/OCA4 | 0.1~0.3 |
| OCA8 | 0.1~0.4(LogMAR)2) |
| OA1 | 0.1~0.4 |
| HPS-11 | 約20/2005) |
各亞型的主要致病基因和特徵如下所示。
| 亞型 | 致病基因 | 主要特徵 |
|---|---|---|
| OCA1 | TYR | 酪胺酸酶缺乏 |
| OCA2 | OCA2(P基因) | 黑素體pH調節1) |
| OCA3 | TYRP1 | 真黑素合成 |
| OCA4 | SLC45A2 | 黑素體轉運蛋白 |
| OCA8 | DCT(TYRP2) | 多巴色素轉化2) |
| OA1 | GPR143 | 黑素體信號3) |
| HPS1/HPS4 | HPS1/HPS4 | BLOC-3複合體4) |
| HPS11 | BLOC1S5 | BLOC-1複合體5) |
OCA2蛋白質的功能:屬於具有12次跨膜α螺旋結構的Na⁺/H⁺反向轉運體家族1)。作為黑素體特異性陰離子通道的組成部分,通過氯離子電流調節I/II期黑素體的pH值1)。ClinVar中登記了477個致病性突變1)。
DCT(TYRP2):催化多巴色素轉化為DHICA(5,6-二羥基吲哚-2-羧酸)2)。是OCA8的致病基因。
GPR143(OA1):調節黑素體囊泡運輸的訊息傳導分子。突變導致大黑素體形成3)。
HPS(BLOC複合體):HPS1/HPS4作為BLOC-3複合體,充當Rab32/38的GEF(鳥嘌呤核苷酸交換因子)4)。BLOC-1由8個亞基組成,參與內體回收;BLOC1S5(BLOS3)基因突變導致HPS-11(2020年首次描述)5)。
由於不同類型在視力預後、全身併發症的有無以及遺傳方式上有所差異,因此準確的亞型診斷非常重要。特別是HPS,常伴有出血傾向、肺纖維化和發炎性腸道疾病,在進行手術或拔牙前需要特別注意。透過基因檢測確定亞型,有助於直接制定適當的管理計劃。
典型的OCA可透過白髮、白皮膚和眼部表現(虹膜透照、眼球震顫)的組合進行臨床診斷。然而,在日本OA1患者中,虹膜中殘留有色素,因此如果僅因眼部症狀就診,診斷可能會延遲。
目前尚無根治性治療方法。治療目標是最大化視覺功能和管理併發症。
視覺管理
眼球震顫與斜視
皮膚與全身管理
防曬:使用防UVB的防曬乳、衣物和帽子保護皮膚。日本人也需注意鱗狀細胞癌的風險。
HPS管理:非類固醇抗發炎藥(包括阿斯匹靈)通常禁忌,因為它們會加重血小板功能障礙。建議從青少年時期開始定期監測肺功能4)。
遺傳諮詢:對於家庭計畫、帶因者診斷和亞型確定至關重要。
對於HPS合併的肺纖維化,使用抗纖維化藥物。
Liu等人(2025)報導,在一例具有新型純合HPS4突變的HPS病例中,尼達尼布使肺纖維化穩定了18個月4)。吡非尼酮也是類似使用的治療選擇。
黑色素合成在黑色素體內透過以下途徑進行:
酪胺酸 → L-多巴 → 多巴醌 → (真黑色素或褐黑色素)
多巴色素轉化為DHICA(5,6-二羥基吲哚-2-羧酸)由DCT(TYRP2)催化2),是真黑色素合成的必需反應。
OCA2黑色素體成熟障礙:OCA2蛋白功能降低導致黑色素體向IV期(成熟黑色素體)的轉變受阻,未成熟的I/II期黑色素體增加1)。透過氯離子電流控制pH調節的破壞是主要機制1)。
當視網膜色素上皮的色素沉著不充分時,發育過程中的光屏蔽作用喪失,阻礙了中心凹的形態發育(無血管區形成和視錐細胞密集遷移)。這是白化症視力不良的根本原因。
在白化症中,來自顳側視網膜的纖維在視交叉處過度交叉到對側,導致比通常更多的纖維被作為鼻側纖維處理。這種錯誤投射可透過VEP檢測為視交叉係數的不對稱2)。
Rateaux等人(2025)的OCA8研究中,使用DCT突變導致L-DOPA降至野生型50%的小鼠模型,顯示L-DOPA補充可恢復眼科異常2)。提示L-DOPA作為黑色素合成中間體在視路形成中發揮作用。
GPR143突變(OA1):GPR143蛋白功能喪失導致黑素體囊泡運輸障礙,形成巨黑素體3)。
HPS(BLOC複合體障礙):BLOC-3功能喪失導致Rab32/38激活障礙,黑素體相關細胞器(LRO:溶酶體相關細胞器)的生物合成全面崩潰4)。血小板內的緻密體和肺II型上皮細胞內的板層小體受到影響。肺纖維化的背景被認為與異常板層小體積累的蠟樣質樣物質有關4)。
BLOC-1(HPS-11):作為包含BLOC1S5(BLOS3)在內的8亞基複合體發揮作用,調控內體循環5)。
L-DOPA是黑色素合成的中間體,同時在視網膜發育和視路形成中發揮重要作用。
在Rateaux等人(2025)的OCA8小鼠模型中,DCT突變導致的L-DOPA降低是眼科表型的主要原因,L-DOPA補充改善了異常2)。正在研究應用於人類6)。
針對白化病患者左旋多巴給藥改善視功能的評估正在進行中,預期具有促進視網膜發育的效果6)。
尼替西農是一種4-羥基苯丙酮酸雙加氧酶(4-HPPD)抑制劑,通過調節酪氨酸代謝途徑可能增加眼內黑色素,正在研究中6)。
針對OCA相關基因(TYR、OCA2等)的基因替代療法之基礎研究正在進行中。Luxturna(RPE65基因替代)獲准用於LCA(萊伯先天性黑矇)加速了遺傳性視網膜疾病整體的基因治療研究6)。
Jiang等人(2024)透過全外顯子組定序(WES)在一個中國家庭中鑑定出OCA2基因的新複合雜合突變(c.635A>G/c.2359+1G>T)1)。功能分析證實,與野生型相比,突變蛋白質向黑色素細胞的遷移受損。
Boeckelmann等人(2021)透過對5例病例(包括2020年後報告的病例)的研究,詳細描述了由BLOC1S5突變引起的HPS-11的臨床譜5)。提供了肺、眼和神經症狀的監測指引。
尼達尼布和吡非尼酮抗纖維化治療對HPS肺纖維化的療效正在持續評估中4)。
Flynn等人(2025)報告,在攜帶GPR143突變的女性OA1攜帶者的眼底檢查中,94%出現弓形視盤前斑片狀色素變化,74%出現虹膜透照缺損3)。研究顯示,即使攜帶者也有很高的眼部異常發生率。
在OCA中,由於缺乏皮膚色素,紫外線引起的DNA損傷容易累積,顯著增加鱗狀細胞癌等皮膚癌的風險。據認為,非洲裔OCA患者的風險尤其高。建議定期皮膚科就診和徹底的防曬(防曬乳、遮蔽衣物)。
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.