Albinismus ist eine Gruppe erblicher Erkrankungen, die durch Genmutationen in der Biosynthese oder dem Transport von Melanin zu einem Mangel oder Fehlen von Pigment in Haut, Haaren und Augen führen. Neben dem Pigmentmangel sind diese Erkrankungen durch spezifische Augen- und Sehbahnabnormalitäten wie Foveahypoplasie, Nystagmus und abnorme Kreuzung der Sehbahn gekennzeichnet.
Es gibt keine kurative Behandlung, und die Sehfunktionsstörung ist nicht fortschreitend (stationär). In Japan ist diese Erkrankung als designierte seltene Krankheit (Seltene-Krankheiten-Gesetz) eingestuft.
Prävalenz: weltweit etwa 1/17.000. Bei Kaukasiern macht OCA1 etwa 50 % aus, bei Afrikanern ist OCA2 am häufigsten (1/10.000)1). Bei chinesischen Han beträgt OCA1 70,1 % und OCA2 10,2 %1).
Okulärer Albinismus (OA)
X-chromosomal-rezessive Vererbung: tritt bei Männern auf. Haut- und Haarpigmentierung normal oder leicht vermindert, Augenbefunde stehen im Vordergrund.
GPR143-Genmutation: beteiligt an der Signaltransduktion der Melanosomen-Biogenese. 192 Mutationen wurden berichtet3).
Konduktorinnen: zeigen ein schlammspritzerartiges mosaikartiges Hypopigmentierungsmuster im Augenhintergrund. Etwa 80 % weisen Netzhautpigmentanomalien auf3).
Syndromaler Albinismus
Hermansky-Pudlak-Syndrom (HPS): OCA + Thrombozytenfunktionsstörung (Blutungsneigung) + Störung der LRO-Biogenese. 11 Subtypen existieren4, 5). Kann mit Lungenfibrose und entzündlichen Darmerkrankungen einhergehen.
Chediak-Higashi-Syndrom (CHS): OCA + Immundefizienz + neurologische Störungen. LYST-Genmutation.
12 Gene: Für den syndromalen Albinismus wurden 12 Gene identifiziert2).
Die weltweite Prävalenz beträgt etwa 1/17.0001), in Europa wird sie mit etwa 1/12.000 angegeben2). Bei Japanern ist eine Verteilung von 34 % OCA1, 27 % OCA4 und 10 % HPS1 bekannt.
QIst Albinismus erblich? Kann er auch auftreten, wenn niemand in der Familie Albinismus hat?
A
OCA wird autosomal-rezessiv vererbt: Wenn beide Eltern jeweils ein mutiertes Allel tragen, beträgt die Wahrscheinlichkeit für das Kind, zu erkranken, 25 %. Da die Eltern phänotypisch normal sein können, ist das Fehlen einer Familienanamnese nicht ungewöhnlich. OA ist X-chromosomal-rezessiv, betrifft hauptsächlich Männer, die Mutter ist Überträgerin. Gentests und genetische Beratung sind wichtig.
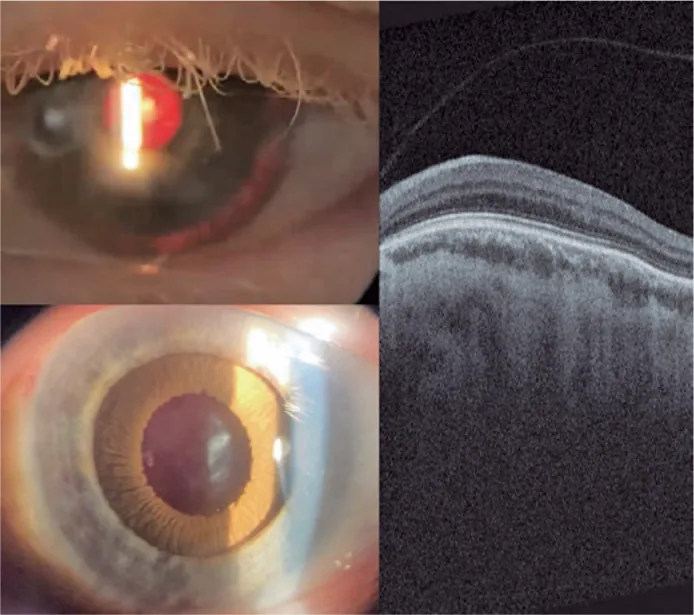
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Spaltlampenfotografie des linken Auges 30 Tage nach der Operation, die die prothetische Iris auf dem Ziliarsulkus bei Retroillumination und nach pharmakologischer Mydriasis zeigt. Netzhaut-OCT zeigt Fehlen der Fovea-Grube und Persistenz der inneren Netzhautschichten im erwarteten Bereich der Fovea (makuläre Hypoplasie).
Foveale Hypoplasie: Wichtigster Faktor für schlechtes Sehen. Die OCT zeigt das Fehlen einer fovealen Vertiefung. In der Fluoreszenzangiographie fehlt häufig die avaskuläre Zone um die Makula. Bei OCA8 zeigen alle Fälle Grad 3, und der Grad korreliert mit der Sehschärfe2).
Iris-Transillumination: Aufgrund des Pigmentmangels der Iris zeigt sich eine erhöhte Transillumination im Spaltlampenmikroskop. Bei OCA8 alle Fälle Grad 32).
Nystagmus (INS): Asymmetrischer, periodisch richtungswechselnder Nystagmus tritt bei etwa 10 % aller INS auf, bei Albinismus-INS bis zu 37 %2).
Fehlleitung der Sehbahn (chiasmal misrouting): Die temporalen Netzhautfasern, die normalerweise ipsilateral (ungekreuzt) verlaufen, kreuzen im Chiasma übermäßig auf die Gegenseite. Nachweis durch VEP. Bei OCA8 wurde ein Chiasma-Koeffizient von −0,97/−0,90 als starke Asymmetrie berichtet2).
Schielen (Strabismus): Bei 53–90,5 % der Fälle vorhanden2).
Hypopigmentierter Fundus: Aufgrund des Pigmentmangels im retinalen Pigmentepithel sind die Aderhautgefäße durchscheinend.
Fundus von OA1-Trägern: 94 % zeigen bandförmige Pigmentveränderungen vor der Arkade, 74 % eine Iris-Transillumination3). In der peripheren Netzhaut können depigmentierte Flecken (Mosaikfundus) auftreten, die zur Identifizierung von Trägern nützlich sind.
QVerändert sich die Sehschärfe bei Albinismus im Laufe des Lebens?
A
Die durch Albinismus verursachte Sehbehinderung ist stationär (nicht fortschreitend) und verschlechtert sich nicht mit dem Alter. Wenn jedoch keine angemessene Refraktionskorrektur oder Amblyopiebehandlung durchgeführt wird, kann die Sehschärfe ohne Verbesserung fixiert bleiben. Eine frühzeitige Intervention mit Refraktionskorrektur und Sehbehindertenversorgung ist wichtig.
Die wichtigsten ursächlichen Gene und Merkmale der einzelnen Subtypen sind unten aufgeführt.
Subtyp
Ursächliches Gen
Hauptmerkmale
OCA1
TYR
Tyrosinase-Mangel
OCA2
OCA2 (P-Gen)
Regulation des Melanosomen-pH1)
OCA3
TYRP1
Eumelanin-Synthese
OCA4
SLC45A2
Melanosomen-Transporter
OCA8
DCT (TYRP2)
Dopachrom-Umwandlung2)
OA1
GPR143
Melanosomen-Signal3)
HPS1/HPS4
HPS1/HPS4
BLOC-3-Komplex4)
HPS11
BLOC1S5
BLOC-1-Komplex5)
Funktion des OCA2-Proteins : Es gehört zur Familie der Na⁺/H⁺-Antiporter mit einer 12-transmembranären α-Helix-Struktur 1). Als Bestandteil eines melanozytenspezifischen Anionenkanals reguliert es den pH-Wert von Melanosomen im Stadium I/II durch Chloridstromkontrolle 1). ClinVar listet 477 pathogene Mutationen 1).
DCT (TYRP2) : Katalysiert die Umwandlung von Dopachrom zu DHICA (5,6-Dihydroxyindol-2-carbonsäure) 2). Es ist das ursächliche Gen für OCA8.
GPR143 (OA1) : Signaltransduktionsmolekül, das den vesikulären Transport von Melanosomen reguliert. Mutationen führen zur Bildung von Makromelanosomen 3).
HPS (BLOC-Komplex) : HPS1/HPS4 fungieren als BLOC-3-Komplex, der als GEF (Guanin-Nukleotid-Austauschfaktor) für Rab32/38 wirkt 4). BLOC-1 besteht aus 8 Untereinheiten und ist am endosomalen Recycling beteiligt; eine Mutation des BLOC1S5 (BLOS3)-Gens verursacht HPS-11 (erstmals 2020 beschrieben) 5).
Blutsverwandtschaft : In Populationen mit häufigen Blutsverwandtschaftsehen, wie in Pakistan, steigt die Prävalenz 4).
Ethnische und regionale Unterschiede : In einigen afrikanischen Regionen ist die Häufigkeit hoch.
Compound-heterozygote Mutationen : Bei OCA2 wurden neue compound-heterozygote Mutationen identifiziert, die zur phänotypischen Vielfalt beitragen 1).
Triallelischer Typ (OCA1) : Kombinationen von drei Allelen des TYR-Gens können zu milder OCA führen 2).
QWarum ist es wichtig, den Typ des Albinismus zu kennen?
A
Da sich Sehprognose, Vorhandensein systemischer Komplikationen und Vererbungsmuster je nach Typ unterscheiden, ist eine genaue Subtyp-Diagnose wichtig. Insbesondere das Hermansky-Pudlak-Syndrom (HPS) geht mit Blutungsneigung, Lungenfibrose und entzündlichen Darmerkrankungen einher, sodass vor Operationen oder Zahnextraktionen Vorsicht geboten ist. Die Identifizierung des Subtyps durch Gentests steht in direktem Zusammenhang mit der Erstellung eines geeigneten Behandlungsplans.
Typischer OCA kann klinisch durch die Kombination von weißen Haaren, weißer Haut und Augenmerkmalen (Iristransillumination, Nystagmus) diagnostiziert werden. Bei japanischen OA1-Patienten bleibt jedoch Pigment in der Iris erhalten, sodass die Diagnose verzögert werden kann, wenn der Patient nur aufgrund von Augensymptomen vorstellig wird.
Spaltlampe : Bestätigung der Iristransillumination. Bewertung des Grades.
OCT (Optische Kohärenztomographie) : Essenziell für das Grading der fovealen Hypoplasie. Auch bei Kindern durchführbar, hilfreich für die Entscheidung zu einer frühen Intervention6).
VEP (visuell evozierte Potenziale) : Am wichtigsten zum Nachweis einer abnormalen Sehbahnkreuzung. Beurteilung der Wellenformasymmetrie zwischen linker und rechter Hemisphäre mittels 3-Kanal-VEP2). Quantifizierung durch den Kreuzungskoeffizienten ( −1 maximale Asymmetrie, +1 normal)2).
Messung des Lambda-Winkels : Ein Lambda-Winkel > 5 Grad ist ein starker klinischer Hinweis auf Albinismus2).
Es gibt keine kurative Behandlung. Ziel der Behandlung ist die Maximierung der Sehfunktion und das Management von Komplikationen.
Visuelles Management
Refraktionskorrektur : Das Tragen einer Brille ab dem Säuglingsalter ist die Grundlage. Eine frühzeitige Intervention zur Verhinderung einer Amblyopie ist wichtig.
Lichtschutzbrille : Verwenden Sie Gläser, die drinnen etwa 20 % und draußen etwa 80 % des Lichts blockieren 5).
Kontaktlinsen mit Iris : Können zur kosmetischen Verbesserung und zur Verringerung der Photophobie verwendet werden.
Sehbehindertenversorgung : Nutzen Sie Hilfsmittel wie Lupen, Bildschirmlesegeräte und Tablets.
Nystagmus und Strabismus
Strabismus-Operation : Wird aus kosmetischen Gründen durchgeführt. Manchmal wird die Dawson-Trick-Litzkow (DTL)-Operation zur Verringerung des Nystagmus durchgeführt.
Nystagmus-Behandlung : Es gibt keine kausale Behandlung. Eine Strabismus-Operation kann zur Korrektur einer abnormalen Kopfhaltung (Nullzone) in Betracht gezogen werden.
Haut- und allgemeine Behandlung
Sonnenschutz : Hautschutz durch UVB-blockierende Sonnencreme, Kleidung und Hüte. Auch bei Japanern ist auf das Risiko eines Plattenepithelkarzinoms zu achten.
HPS-Management : NSAR (einschließlich Aspirin) sind in der Regel kontraindiziert, da sie die Thrombozytenfunktion verschlechtern. Eine regelmäßige Überwachung der Lungenfunktion ab dem Teenageralter wird empfohlen 4).
Genetische Beratung : Essenziell für Familienplanung, Trägerdiagnostik und Subtypbestimmung.
Für die mit HPS assoziierte Lungenfibrose werden antifibrotische Medikamente eingesetzt.
Liu et al. (2025) berichteten über einen HPS-Fall mit einer neuen homozygoten HPS4-Mutation, bei dem die Gabe von Nintedanib die Lungenfibrose für 18 Monate stabilisierte 4). Pirfenidon ist eine ebenfalls verwendete Behandlungsoption.
QIst die Gentherapie für Albinismus bereits in der klinischen Praxis verfügbar?
A
Derzeit ist die Gentherapie für Albinismus noch nicht in der klinischen Praxis verfügbar. Für andere erbliche Netzhauterkrankungen wie Retinoschisis und LCA sind Gentherapien zugelassen (z. B. Luxturna), und die Forschung zur Anwendung bei Albinismus schreitet voran6). Derzeit stehen Refraktionskorrektur und Low-Vision-Versorgung im Mittelpunkt der Standardbehandlung.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Die Melaninsynthese verläuft in den Melanosomen über folgenden Weg:
Tyrosin → L-DOPA → DOPA-Chinon → (Eumelanin oder Phäomelanin)
Die Umwandlung von Dopachrom zu DHICA (5,6-Dihydroxyindol-2-carbonsäure) wird durch DCT (TYRP2) katalysiert2), eine für die Eumelaninsynthese essentielle Reaktion.
Melanosomen-Reifungsstörung bei OCA2 : Die verminderte Funktion des OCA2-Proteins führt zu einer gestörten Transition der Melanosomen in das Stadium IV (reife Melanosomen) und einer Zunahme unreifer Melanosomen der Stadien I/II1). Der Hauptmechanismus ist eine Störung der pH-Regulation durch die Kontrolle des Chloridstroms1).
Wenn die Pigmentierung des retinalen Pigmentepithels unzureichend ist, geht die lichtabschirmende Wirkung während der Entwicklung verloren, was die morphologische Entwicklung der Fovea (Bildung der avaskulären Zone und dichte Migration der Zapfen) behindert. Dies ist die grundlegende Ursache für die schlechte Sehschärfe bei Albinismus.
Bei Albinismus kreuzen die Fasern aus der temporalen Netzhaut im Chiasma übermäßig auf die Gegenseite, sodass mehr Fasern als nasal verarbeitet werden. Dieses „Misrouting“ wird im VEP als Asymmetrie des Chiasma-Koeffizienten erfasst2).
In ihrer OCA8-Studie (2025) verwendeten Rateaux et al. ein Mausmodell, bei dem eine DCT-Mutation den L-DOPA-Spiegel auf 50 % des Wildtyps senkt, und zeigten, dass eine L-DOPA-Supplementierung die ophthalmologischen Anomalien korrigierte 2). Dies deutet darauf hin, dass L-DOPA als Zwischenprodukt der Melaninsynthese eine Rolle bei der Bildung der Sehbahn spielt.
GPR143-Mutation (OA1) : Der Funktionsverlust des GPR143-Proteins beeinträchtigt den vesikulären Transport von Melanosomen, was zur Bildung von Makromelanosomen führt 3).
HPS (BLOC-Komplex-Störungen) : Der Funktionsverlust von BLOC-3 beeinträchtigt die Aktivierung von Rab32/38, was zu einer allgemeinen Störung der Biogenese von Melanosomen-assoziierten Organellen (LRO: lysosomenverwandte Organellen) führt 4). Dichte Körperchen in Thrombozyten und Lamellarkörperchen in Lungenepithelzellen Typ II sind betroffen. Hinter der Lungenfibrose wird eine Ansammlung von ceroidähnlichen Substanzen aus abnormalen Lamellarkörperchen vermutet 4).
BLOC-1 (HPS-11) : Funktionert als 8-Untereinheiten-Komplex, der BLOC1S5 (BLOS3) umfasst, und reguliert das endosomale Recycling 5).
7. Aktuelle Forschung und Zukunftsperspektiven (Forschungsstadium)
L-DOPA ist ein Zwischenprodukt der Melaninsynthese und spielt eine wichtige Rolle bei der Netzhautentwicklung und der Bildung der Sehbahn.
Im OCA8-Mausmodell von Rateaux et al. (2025) war der durch die DCT-Mutation verursachte L-DOPA-Abfall der Hauptfaktor für den ophthalmologischen Phänotyp, und die L-DOPA-Supplementierung verbesserte die Anomalien 2). Eine Anwendung beim Menschen wird erforscht 6).
Die Bewertung der Verbesserung der Sehfunktion durch Levodopa-Gabe bei Albinismus-Patienten läuft, und es wird ein fördernder Effekt auf die Netzhautentwicklung erwartet 6).
Nitisinon, ein Inhibitor der 4-Hydroxyphenylpyruvat-Dioxygenase (4-HPPD), wird auf sein Potenzial untersucht, durch Regulierung des Tyrosin-Stoffwechselwegs das intraokulare Melanin zu erhöhen 6).
Die Grundlagenforschung zur Gentherapie, die auf OCA-assoziierte Gene (TYR, OCA2 usw.) abzielt, schreitet voran. Die Zulassung von Luxturna (RPE65-Genersatz) für LCA (Lebersche kongenitale Amaurose) beschleunigt die Gentherapieforschung für erbliche Netzhauterkrankungen im Allgemeinen 6).
Jiang et al. (2024) identifizierten durch Whole-Exome-Sequenzierung (WES) in einer chinesischen Familie eine neue compound-heterozygote Mutation (c.635A>G/c.2359+1G>T) des OCA2-Gens 1). Die Funktionsanalyse bestätigte, dass das mutierte Protein im Vergleich zum Wildtyp in seiner Translokation zu Melanozyten beeinträchtigt ist.
Boeckelmann et al. (2021) beschrieben das klinische Spektrum des durch BLOC1S5-Mutation verursachten HPS-11 in einer Studie mit 5 Fällen, einschließlich seit 2020 berichteter Fälle 5). Es werden Leitlinien zur Überwachung von Lungen-, Augen- und neurologischen Symptomen gegeben.
Die Wirksamkeit der antifibrotischen Therapie mit Nintedanib und Pirfenidon bei HPS-Lungenfibrose wird kontinuierlich evaluiert 4).
Flynn et al. (2025) berichteten, dass bei der Fundusuntersuchung von OA1-Trägerinnen mit GPR143-Mutation bei 94% bogenförmige makuläre Pigmentveränderungen und bei 74% eine Iris-Transillumination festgestellt wurden 3). Es wurde gezeigt, dass auch bei Trägerinnen ophthalmologische Auffälligkeiten häufig auftreten.
QHaben Patienten mit Albinismus ein erhöhtes Hautkrebsrisiko?
A
Bei OCA führt das Fehlen von Hautpigment zu einer leichteren Anhäufung von UV-bedingten DNA-Schäden, was das Risiko für Hautkrebs wie Plattenepithelkarzinome signifikant erhöht. Bei OCA-Patienten afrikanischer Abstammung wird das Risiko als besonders hoch eingeschätzt. Regelmäßige dermatologische Untersuchungen und konsequenter Sonnenschutz (Sonnencreme, bedeckende Kleidung) werden empfohlen.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.