آلبینیسم (Albinism) گروهی از بیماریهای ارثی است که در اثر جهش در ژنهای دخیل در بیوسنتز یا انتقال رنگدانه ملانین، باعث فقدان یا کاهش رنگدانه در پوست، مو و چشم میشود. ویژگی این گروه از بیماریها علاوه بر کمبود رنگدانه، وجود ناهنجاریهای خاص چشمی و بینایی مانند دیسپلازی فووآ، نیستاگموس و تقاطع غیرطبیعی مسیر بینایی است.
درمان قطعی وجود ندارد و اختلال بینایی غیرپیشرونده (پایدار) است. در ژاپن به عنوان بیماری نادر (طبق قانون بیماریهای نادر) تعیین شده است.
وراثت اتوزومال مغلوب: شایعترین گروه. باعث کمبود رنگدانه در پوست، مو و چشم میشود.
8 ژن و 8 زیرگروه: OCA1 (TYR)، OCA2 (ژن OCA2/P)، OCA3 (TYRP1)، OCA4 (SLC45A2)، OCA8 (DCT) و غیره.
شیوع: حدود 1 در 17,000 در جهان. در سفیدپوستان، OCA1 حدود 50٪ را تشکیل میدهد و در آفریقاییتباران، OCA2 شایعترین است (1 در 10,000)1). در قوم هان چین، OCA1 70.1٪ و OCA2 10.2٪ است1).
آلبینیسم چشمی (OA)
وراثت وابسته به X مغلوب: در مردان بروز میکند. رنگدانه پوست و مو طبیعی یا کمی کاهش یافته و یافتههای چشمی غالب هستند.
جهش در ژن GPR143: در انتقال سیگنال بیوسنتز ملانوزوم نقش دارد. 192 جهش گزارش شده است3).
زنان ناقل: الگوی موزاییکی هیپوپیگمانتاسیون شبیه گلپاشی در فوندوس نشان میدهند. حدود 80٪ ناهنجاری رنگدانه شبکیه دارند3).
آلبینیسم سندرمیک
سندرم هرمنسکی-پودلاک (HPS): OCA + اختلال عملکرد پلاکت (تمایل به خونریزی) + اختلال بیوسنتز LRO. 11 زیرگروه وجود دارد4, 5). ممکن است با فیبروز ریوی و بیماری التهابی روده همراه باشد.
سندرم چدیاک-هیگاشی (CHS): OCA + نقص ایمنی + اختلال عصبی. جهش در ژن LYST.
12 ژن: برای آلبینیسم سندرمیک 12 ژن شناسایی شده است2).
شیوع جهانی حدود 1 در 17,000 است1) و در اروپا حدود 1 در 12,000 گزارش شده است2). در ژاپنیها، توزیع OCA1 34٪، OCA4 27٪ و HPS1 10٪ شناخته شده است.
Qآیا آلبینیسم ارثی است؟ آیا حتی اگر در خانواده فرد مبتلا به آلبینیسم نباشد، ممکن است بروز کند؟
A
OCA یک بیماری اتوزومال مغلوب است و اگر هر دو والد ناقل یک آلل جهشیافته باشند، با احتمال ۲۵٪ کودک مبتلا میشود. از آنجا که ممکن است والدین از نظر فنوتیپ طبیعی باشند اما فرزند مبتلا شود، عدم وجود سابقه خانوادگی غیرمعمول نیست. OA یک بیماری مغلوب وابسته به X است که عمدتاً مردان را مبتلا میکند و مادر ناقل است. آزمایش ژنتیک و مشاوره ژنتیک مهم هستند.
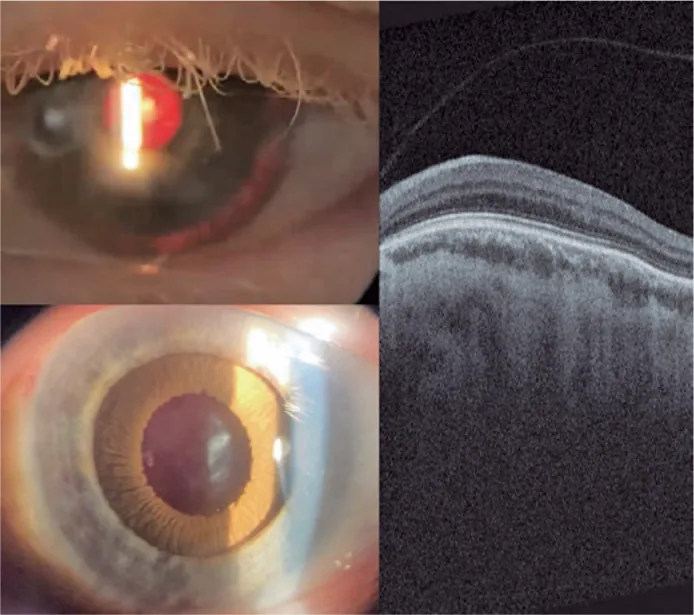
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
عکس با لامپ شکاف چشم چپ ۳۰ روز پس از جراحی، نشاندهنده عنبیه مصنوعی روی شیار مژگانی از طریق روشنایی معکوس و پس از گشاد شدن دارویی مردمک. OCTشبکیه نشاندهنده عدم وجود حفره مرکزی و تداوم لایههای داخلی شبکیه در ناحیه مورد انتظار حفره (هیپوپلازی ماکولا).
ناهنجاری حفره مرکزی (foveal hypoplasia): مهمترین عامل بینایی ضعیف. در OCT فقدان فرورفتگی حفره مرکزی تأیید میشود. در آنژیوگرافی فلورسئین، عدم وجود ناحیه بدون عروق اطراف ماکولا نیز یافته مشخصهای است. در OCA8 همه موارد درجه ۳ دارند و درجه با بینایی همبستگی دارد 2).
شفافیت عنبیه (iris transillumination): به دلیل فقدان رنگدانه عنبیه، در معاینه با لامپ شکاف افزایش شفافیت دیده میشود. در OCA8 همه موارد درجه ۳ دارند 2).
نیستاگموس (INS): نیستاگموس متناوب نامتقارن در حدود ۱۰٪ از کل موارد INS و تا ۳۷٪ در آلبینیسم INS دیده میشود 2).
مسیر بینایی غیرطبیعی (chiasmal misrouting): فیبرهای شبکیه تمپورال که باید مسیر همطرف (uncrossed) را طی کنند، در کیاسمای بینایی بیش از حد به طرف مقابل عبور میکنند. با VEP تشخیص داده میشود. در OCA8 ضریب کیاسمایی ۰٫۹۷-/۰٫۹۰- با عدم تقارن شدید گزارش شده است 2).
استرابیسم (انحراف چشم): در ۵۳ تا ۹۰٫۵٪ موارد دیده میشود 2).
فوندوس هیپوپیگمانته: به دلیل فقدان رنگدانه در اپیتلیوم رنگدانه شبکیه، عروق کوروئید قابل مشاهده هستند.
فوندوس ناقلان OA1: در ۹۴٪ تغییرات رنگدانهای لکهای در جلوی قوسهای عروقی و در ۷۴٪ شفافیت عنبیه دیده میشود 3). ممکن است لکههای دپیگمانته در شبکیه محیطی (فوندوس موزائیک) دیده شود که برای شناسایی ناقلان مفید است.
Qآیا بینایی در آلبینیسم در طول زندگی تغییر میکند؟
A
اختلال بینایی ناشی از آلبینیسم ایستا (غیر پیشرونده) است و با افزایش سن بدتر نمیشود. با این حال، اگر اصلاح عیوب انکساری یا درمان تنبلی چشم به درستی انجام نشود، ممکن است بینایی بدون بهبود ثابت بماند. مداخله زودهنگام با اصلاح عیوب انکساری و مراقبتهای کمبینایی مهم است.
عملکرد پروتئین OCA2: متعلق به خانواده آنتیپورتر Na⁺/H⁺ با ساختار ۱۲ مارپیچ آلفای تراغشایی است 1). به عنوان جزئی از کانال آنیون اختصاصی ملانوزوم، pH ملانوزومهای مرحله I/II را از طریق تنظیم جریان کلرید کنترل میکند 1). ۴۷۷ جهش بیماریزا در ClinVar ثبت شده است 1).
DCT (TYRP2): تبدیل دوپاکروم به DHICA (۵،۶-دیهیدروکسیایندول-۲-کربوکسیلیک اسید) را کاتالیز میکند 2). ژن عامل OCA8 است.
GPR143 (OA1): مولکول انتقال پیام که انتقال وزیکولی ملانوزوم را تنظیم میکند. جهش آن منجر به تشکیل ماکروملانوزوم میشود 3).
HPS (کمپلکس BLOC): HPS1/HPS4 به عنوان کمپلکس BLOC-3 به عنوان GEF (عامل تبادل نوکلئوتید گوانین) برای Rab32/38 عمل میکنند 4). BLOC-1 از ۸ زیرواحد تشکیل شده و در بازیافت اندوزومی نقش دارد و جهش در ژن BLOC1S5 (BLOS3) باعث HPS-11 (اولین بار در سال ۲۰۲۰ توصیف شد) میشود 5).
ازدواج فامیلی: در جمعیتهایی با ازدواج فامیلی بالا مانند پاکستان، شیوع افزایش مییابد 4).
تفاوت نژادی و منطقهای: در برخی مناطق آفریقا فراوانی بالاست.
جهشهای هتروزیگوت مرکب: در OCA2 جهشهای هتروزیگوت مرکب جدید شناسایی شده که در تنوع فنوتیپی نقش دارند 1).
نوع سه آللی (OCA1): ترکیب سه آلل در ژن TYR میتواند باعث OCA خفیف شود 2).
Qچرا دانستن نوع آلبینیسم اهمیت دارد؟
A
از آنجایی که پیشآگهی بینایی، وجود عوارض سیستمیک و الگوی وراثت بر اساس نوع متفاوت است، تشخیص دقیق زیرگروه اهمیت دارد. به ویژه در HPS، تمایل به خونریزی، فیبروز ریوی و بیماری التهابی روده دیده میشود و قبل از جراحی یا کشیدن دندان باید احتیاط کرد. شناسایی زیرگروه با آزمایش ژنتیک مستقیماً به برنامهریزی مدیریت مناسب منجر میشود.
OCA معمولی را میتوان از طریق ترکیب موی سفید، پوست سفید و یافتههای چشمی (شفافیت عنبیه، نیستاگموس) به صورت بالینی تشخیص داد. با این حال، در OA1 ژاپنی، رنگدانه در عنبیه باقی میماند، بنابراین اگر بیمار فقط با علائم چشمی مراجعه کند، تشخیص ممکن است به تأخیر بیفتد.
لامپ اسلیت (بیومیکروسکوپ) : تأیید شفافیت عنبیه. ارزیابی درجهبندی انجام میشود.
OCT (توموگرافی انسجام نوری) : برای درجهبندی هیپوپلازی فووآ ضروری است. حتی در کودکان نیز قابل انجام است و به تصمیمگیری برای مداخله زودهنگام کمک میکند 6).
VEP (پتانسیل برانگیخته بینایی) : برای تشخیص عبور غیرطبیعی مسیر بینایی بسیار مهم است. با استفاده از VEP سه لید، عدم تقارن شکل موج بین نیمکرههای چپ و راست ارزیابی میشود 2). با ضریب کیاسمای بینایی (1- حداکثر عدم تقارن، 1+ طبیعی) کمّی میشود 2).
اندازهگیری زاویه لامبدا : زاویه لامبدا بیشتر از 5 درجه یک شاخص بالینی قوی برای آلبینیسم است 2).
آنژیوگرافی فلورسئین (FA) : الگوی موزاییکی فوندوس را در ناقلان OA1 واضحتر میکند.
میکروسکوپ الکترونی پلاکت : برای تشخیص قطعی HPS استفاده میشود. فقدان گرانولهای متراکم (dense body) یک یافته مشخص است 4).
آزمایش ژنتیک : توالییابی کامل اگزوم (WES) 1) یا توالییابی پانلی 2, 4, 5). به ویژه در مواردی که فنوتیپ خفیف یا غیر معمول است، اهمیت دارد.
درمان قطعی وجود ندارد. هدف درمان، به حداکثر رساندن عملکرد بینایی و مدیریت عوارض است.
مدیریت بینایی
اصلاح عیوب انکساری: اصلاح با عینک از دوران نوزادی و کودکی اساس درمان است. مداخله زودهنگام برای پیشگیری از تنبلی چشم اهمیت دارد.
عینکهای محافظ نور: در داخل خانه از لنزهایی با حدود ۲۰٪ و در خارج از خانه از لنزهایی با حدود ۸۰٪ محافظت در برابر نور استفاده میشود5).
لنزهای تماسی رنگی: میتوان برای بهبود ظاهر و کاهش حساسیت به نور استفاده کرد.
مراقبت از کمبینایی: استفاده از وسایل کمکی مانند ذرهبین، ذرهبین مطالعه و دستگاههای تبلت.
نیستاگموس و استرابیسم
جراحی استرابیسم: برای اهداف زیبایی انجام میشود. در برخی موارد، جراحی Dawson-Trick-Litzkow (DTL) برای کاهش نیستاگموس انجام میشود.
درمان نیستاگموس: درمان قطعی وجود ندارد. برای مقابله با وضعیت غیرطبیعی سر (برای حفظ ناحیه صفر)، جراحی استرابیسم در نظر گرفته میشود.
مدیریت پوست و سیستمیک
محافظت در برابر نور خورشید: محافظت از پوست با کرمهای ضدآفتاب مسدودکننده UVB، لباس و کلاه. حتی در ژاپنیها نیز باید مراقب خطر کارسینوم سلول سنگفرشی بود.
مدیریت HPS: NSAIDs (از جمله آسپرین) به دلیل تشدید اختلال عملکرد پلاکتی معمولاً منع مصرف دارند. پایش منظم عملکرد ریه از دهه دوم زندگی توصیه میشود4).
مشاوره ژنتیک: برای برنامهریزی خانواده، تشخیص ناقل و تعیین زیرگروه ضروری است.
برای فیبروز ریوی همراه با HPS از داروهای ضد فیبروز استفاده میشود.
Liu و همکاران (2025) در یک مورد HPS با جهش جدید هموزیگوت HPS4، تثبیت فیبروز ریوی به مدت ۱۸ ماه را پس از تجویز نینتادانیب گزارش کردند4). پیرفنیدون نیز یک گزینه درمانی مشابه است.
Qآیا ژن درمانی برای آلبینیسم عملی شده است؟
A
در حال حاضر ژن درمانی برای آلبینیسم عملی نشده است. در سایر بیماریهای ارثی شبکیه مانند رتینوشیزیس و LCA، ژن درمانی تأیید شده است (مثلاً Luxturna) و تحقیقات برای کاربرد آن در آلبینیسم در حال پیشرفت است6). در حال حاضر، اصلاح عیوب انکساری و مراقبت از کمبینایی، محور درمان استاندارد هستند.
سنتز ملانین در ملانوزومها از مسیر زیر پیروی میکند:
تیروزین → L-DOPA → دوپاکینون → (اوملانین یا فئوملانین)
تبدیل دوپاکروم به DHICA (5،6-دیهیدروکسیایندول-2-کربوکسیلیک اسید) توسط DCT (TYRP2) کاتالیز میشود2) و واکنشی ضروری برای سنتز اوملانین است.
اختلال بلوغ ملانوزوم در OCA2: کاهش عملکرد پروتئین OCA2 باعث اختلال در انتقال ملانوزومها به مرحله IV (ملانوزوم بالغ) و افزایش ملانوزومهای نابالغ مرحله I/II میشود1). مکانیسم اصلی، اختلال در تنظیم pH از طریق کنترل جریان کلر است1).
هنگامی که رنگدانهگذاری اپیتلیوم رنگدانهای شبکیه نامناسب باشد، اثر محافظتی نور در طول تکامل از بین میرود و رشد مورفولوژیکی فووئا (تشکیل ناحیه بدون عروق و تراکم سلولهای مخروطی) مختل میشود. این علت اصلی ضعف بینایی در آلبینیسم است.
در آلبینیسم، فیبرهای شبکیه تمپورال در کیاسمای بینایی بیش از حد به طرف مقابل تقاطع میکنند و تعداد بیشتری فیبر نسبت به حالت عادی به عنوان فیبرهای بینی پردازش میشوند. این «مسیریابی اشتباه» به صورت عدم تقارن ضریب کیاسمایی در VEP قابل تشخیص است2).
در مطالعه OCA8 توسط Rateaux و همکاران (2025)، با استفاده از مدل موشی که در آن جهش DCT باعث کاهش L-DOPA به 50% نوع وحشی میشود، نشان داده شد که تجویز L-DOPA ناهنجاریهای چشمی را بهبود میبخشد 2). این یافته نقش L-DOPA را به عنوان یک واسطه در سنتز ملانین در شکلگیری مسیر بینایی نشان میدهد.
جهش GPR143 (OA1): از دست دادن عملکرد پروتئین GPR143 باعث اختلال در انتقال وزیکولی ملانوزومها و تشکیل ماکروملانوزومها میشود 3).
HPS (اختلال کمپلکس BLOC): از دست دادن عملکرد BLOC-3 باعث اختلال در فعالسازی Rab32/38 و شکست کلی در بیوسنتز اندامکهای مرتبط با ملانوزوم (LRO) میشود 4). اجسام متراکم در پلاکتها و لاملار bodies در سلولهای اپیتلیال نوع II ریه تحت تأثیر قرار میگیرند. زمینه فیبروز ریوی با تجمع مواد شبه سروئید از لاملار bodies غیرطبیعی مرتبط است 4).
BLOC-1 (HPS-11): به عنوان یک کمپلکس ۸ زیرواحدی شامل BLOC1S5 (BLOS3) عمل میکند و بازیافت اندوزومی را تنظیم میکند 5).
7. تحقیقات جدید و چشمانداز آینده (گزارشهای مرحله تحقیقاتی)
L-DOPA یک واسطه در سنتز ملانین است و همچنین نقش مهمی در رشد شبکیه و شکلگیری مسیر بینایی ایفا میکند.
در مدل موشی OCA8 توسط Rateaux و همکاران (2025)، کاهش L-DOPA ناشی از جهش DCT عامل اصلی فنوتیپ چشمی بود و تجویز L-DOPA ناهنجاریها را بهبود بخشید 2). کاربرد آن در انسان در حال بررسی است 6).
ارزیابی بهبود عملکرد بینایی با تجویز لوودوپا در بیماران مبتلا به آلبینیسم در حال انجام است و اثرات تقویت رشد شبکیه مورد انتظار است 6).
نیتیسینون، یک مهارکننده ۴-هیدروکسیفنیلپیروات دیاکسیژناز (4-HPPD)، با تنظیم مسیر متابولیسم تیروزین، احتمال افزایش ملانین داخل چشمی را مورد مطالعه قرار میدهد 6).
تحقیقات پایهای در زمینه ژن درمانی جایگزینی با هدف قرار دادن ژنهای مرتبط با OCA (TYR، OCA2 و غیره) در حال پیشرفت است. تأیید Luxturna (جایگزینی ژن RPE65) برای LCA (آموروز مادرزادی لبر) تحقیقات ژن درمانی برای بیماریهای ارثی شبکیه را به طور کلی تسریع کرده است 6).
Jiang و همکاران (2024) با استفاده از توالییابی کامل اگزوم (WES) در یک خانواده چینی، یک جهش هتروزیگوت مرکب جدید (c.635A>G/c.2359+1G>T) در ژن OCA2 شناسایی کردند 1). تجزیه و تحلیل عملکردی تأیید کرد که پروتئین جهشیافته در مقایسه با نوع وحشی، انتقال به ملانوسیتها را مختل میکند.
Boeckelmann و همکاران (2021) با بررسی 5 مورد شامل موارد گزارش شده از سال 2020 به بعد، طیف بالینی سندرم HPS-11 ناشی از جهش BLOC1S5 را به تفصیل شرح دادند 5). دستورالعملهایی برای پایش علائم ریوی، چشمی و عصبی ارائه شده است.
اثربخشی درمان ضد فیبروز با نینتادانیب و پیرفنیدون برای فیبروز ریوی HPS به طور مداوم در حال ارزیابی است 4).
Flynn و همکاران (2025) گزارش کردند که در معاینه فوندوس زنان ناقل OA1 با جهش GPR143، 94% تغییرات رنگدانهای لکهای قوسی شکل قدامی و 74% ترانسایلومیناسیون عنبیه مشاهده شد 3). نشان داده شد که ناهنجاریهای چشمی حتی در ناقلان نیز با شیوع بالایی رخ میدهد.
Qآیا بیماران مبتلا به آلبینیسم در معرض خطر بالای سرطان پوست هستند؟
A
در OCA به دلیل فقدان رنگدانه پوست، آسیب DNA ناشی از اشعه ماوراء بنفش به راحتی تجمع مییابد و خطر سرطان پوست مانند کارسینوم سلول سنگفرشی به طور قابل توجهی افزایش مییابد. این خطر به ویژه در بیماران OCA آفریقایی تبار بالا است. مراجعه منظم به متخصص پوست و محافظت کامل در برابر آفتاب (کرم ضد آفتاب، لباس پوشاننده) توصیه میشود.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.