Bạch tạng (Albinism) là một nhóm bệnh di truyền do đột biến gen liên quan đến sinh tổng hợp hoặc vận chuyển sắc tố melanin, dẫn đến thiếu hoặc giảm sắc tố ở da, tóc và mắt. Ngoài thiếu sắc tố, bệnh còn có các bất thường đặc trưng ở mắt và đường thị giác như thiểu sản hố trung tâm, rung giật nhãn cầu và bắt chéo bất thường của đường thị giác.
Không có phương pháp điều trị triệt để, và suy giảm chức năng thị giác là không tiến triển (tĩnh). Tại Nhật Bản, bệnh được chỉ định là bệnh hiếm gặp cụ thể (theo Luật Bệnh hiếm).
Bệnh bạch tạng được phân loại thành ba nhóm chính.
Bạch tạng da-mắt (OCA)
Di truyền lặn nhiễm sắc thể thường: Nhóm phổ biến nhất. Gây thiếu hụt sắc tố ở da, tóc và mắt.
8 gen và 8 phân nhóm: OCA1 (TYR), OCA2 (gen OCA2/P), OCA3 (TYRP1), OCA4 (SLC45A2), OCA8 (DCT) và các gen khác.
Tỷ lệ mắc: Khoảng 1/17.000 trên toàn thế giới. Ở người da trắng, OCA1 chiếm khoảng 50%; ở người gốc Phi, OCA2 phổ biến nhất (1/10.000)1). Ở người Hán Trung Quốc, OCA1 chiếm 70,1% và OCA2 chiếm 10,2%1).
Bạch tạng mắt (OA)
Di truyền lặn liên kết X: Xảy ra ở nam giới. Sắc tố da và tóc bình thường hoặc giảm nhẹ, với các biểu hiện ở mắt là chủ yếu.
Đột biến gen GPR143: Liên quan đến truyền tín hiệu sinh tổng hợp melanosome. Đã có 192 đột biến được báo cáo3).
Phụ nữ mang gen: Có dạng giảm sắc tố khảm giống vết bùn bắn ở đáy mắt. Khoảng 80% có bất thường sắc tố võng mạc3).
Bạch tạng hội chứng
Hội chứng Hermansky-Pudlak (HPS): OCA + rối loạn chức năng tiểu cầu (xu hướng chảy máu) + rối loạn sinh tổng hợp LRO. Có 11 phân nhóm4, 5). Có thể kèm theo xơ phổi và bệnh viêm ruột.
Hội chứng Chediak-Higashi (CHS): OCA + suy giảm miễn dịch + rối loạn thần kinh. Đột biến gen LYST.
12 gen: 12 gen đã được xác định cho bạch tạng hội chứng2).
Tỷ lệ mắc trên thế giới khoảng 1/17.0001), và ở châu Âu được báo cáo khoảng 1/12.0002). Ở người Nhật, phân bố được biết đến là OCA1 34%, OCA4 27%, HPS1 10%.
QBệnh bạch tạng có di truyền không? Có thể mắc bệnh ngay cả khi không có ai trong gia đình bị bạch tạng không?
A
OCA là bệnh di truyền lặn trên nhiễm sắc thể thường, nếu cả bố và mẹ đều mang một alen đột biến (người mang gen), xác suất con mắc bệnh là 25%. Bệnh có thể xảy ra ngay cả khi bố mẹ có kiểu hình bình thường, do đó không hiếm gặp dù không có tiền sử gia đình. OA là bệnh lặn liên kết X, chủ yếu ảnh hưởng đến nam giới, mẹ là người mang gen. Xét nghiệm di truyền và tư vấn di truyền rất quan trọng.
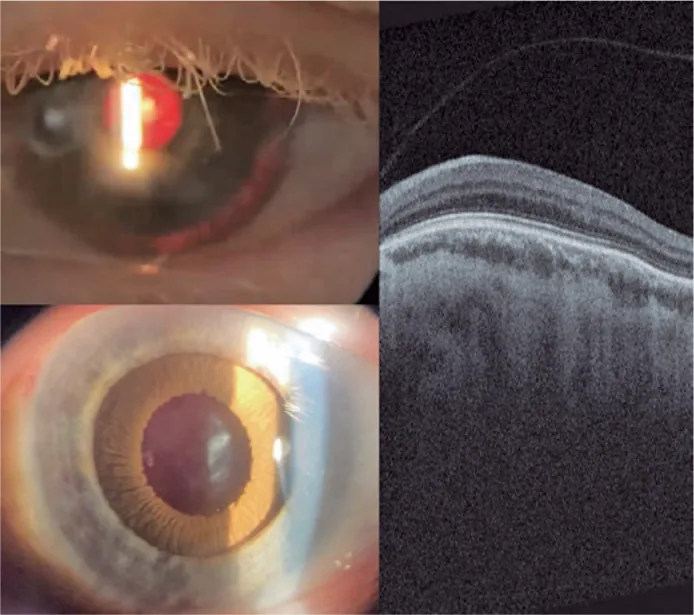
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Ảnh chụp đèn khe mắt trái 30 ngày sau phẫu thuật, cho thấy mống mắt giả trên sulcus thể mi qua chiếu sáng ngược và sau khi giãn đồng tử bằng thuốc. OCTvõng mạc cho thấy không có hố trung tâm và sự tồn tại của các lớp võng mạc bên trong qua vùng dự kiến của hố trung tâm (thiểu sản hoàng điểm).
Thiểu sản hố trung tâm: Yếu tố quan trọng nhất gây thị lực kém. Sự vắng mặt của lõm hố trung tâm được xác nhận bằng OCT. Trên chụp mạch huỳnh quang, vùng vô mạch quanh hố thường không thấy. Ở OCA8, tất cả các trường hợp đều ở độ 3, và độ tương quan với thị lực2).
Xuyên sáng mống mắt: Do thiếu sắc tố mống mắt, thấy tăng xuyên sáng khi khám bằng đèn khe. Ở OCA8, tất cả các trường hợp đều độ 3 2).
Bất thường bắt chéo đường thị giác (chiasmal misrouting): Các sợi võng mạc thái dương, lẽ ra phải đi theo đường cùng bên (không bắt chéo), lại bắt chéo quá mức sang bên đối diện tại giao thoa thị giác. Được phát hiện bằng VEP. Ở OCA8, hệ số giao thoa được báo cáo là -0,97/-0,90, cho thấy sự bất đối xứng nặng 2).
Lác: Gặp ở 53-90,5% trường hợp 2).
Đáy mắt giảm sắc tố: Do thiếu sắc tố biểu mô sắc tố võng mạc, các mạch mạch máu màng bồ đào có thể nhìn thấy.
Đáy mắt của người mang gen OA1: 94% có thay đổi sắc tố dạng mảng trước các mạch hình vòng cung, 74% có xuyên sáng mống mắt3). Có thể thấy các đốm giảm sắc tố ở võng mạc ngoại vi (đáy mắt khảm), hữu ích để xác định người mang gen.
QThị lực ở bệnh bạch tạng có thay đổi suốt đời không?
A
Rối loạn thị giác do bạch tạng là tĩnh (không tiến triển) và không xấu đi theo tuổi tác. Tuy nhiên, nếu không được điều chỉnh khúc xạ thích hợp hoặc điều trị nhược thị, thị lực có thể vẫn kém mà không cải thiện. Can thiệp sớm bằng điều chỉnh khúc xạ và chăm sóc thị lực kém là rất quan trọng.
Dưới đây là các gen gây bệnh chính và đặc điểm của từng phân nhóm.
Phân nhóm
Gen gây bệnh
Đặc điểm chính
OCA1
TYR
Thiếu hụt tyrosinase
OCA2
OCA2 (gen P)
Điều hòa pH melanosome1)
OCA3
TYRP1
Tổng hợp eumelanin
OCA4
SLC45A2
Chất vận chuyển melanosome
OCA8
DCT (TYRP2)
Chuyển đổi dopachrome 2)
OA1
GPR143
Tín hiệu melanosome 3)
HPS1/HPS4
HPS1/HPS4
Phức hợp BLOC-3 4)
HPS11
BLOC1S5
Phức hợp BLOC-1 5)
Chức năng của protein OCA2: Thuộc họ antiporter Na⁺/H⁺ với cấu trúc xoắn α xuyên màng 12 lần 1). Là thành phần của kênh anion đặc hiệu melanosome, điều chỉnh pH của melanosome giai đoạn I/II thông qua kiểm soát dòng chloride 1). Có 477 đột biến gây bệnh được đăng ký trong ClinVar 1).
DCT (TYRP2): Xúc tác chuyển đổi dopachrome thành DHICA (axit 5,6-dihydroxyindole-2-carboxylic) 2). Là gen gây bệnh OCA8.
GPR143 (OA1): Phân tử tín hiệu kiểm soát vận chuyển túi melanosome. Đột biến dẫn đến hình thành melanosome khổng lồ 3).
HPS (Phức hợp BLOC): HPS1/HPS4 hoạt động như phức hợp BLOC-3 với vai trò GEF (yếu tố trao đổi nucleotide guanine) cho Rab32/38 4). BLOC-1 gồm 8 tiểu đơn vị và tham gia vào tái chế nội bào, đột biến gen BLOC1S5 (BLOS3) gây ra HPS-11 (được mô tả lần đầu năm 2020) 5).
Hôn nhân cận huyết: Tỷ lệ mắc bệnh tăng cao trong các quần thể có tỷ lệ kết hôn cận huyết cao như Pakistan 4).
Khác biệt chủng tộc và địa lý: Tần suất cao ở một số vùng châu Phi.
Đột biến dị hợp tử kép (compound heterozygous): Các đột biến dị hợp tử kép mới đã được xác định ở OCA2, góp phần vào sự đa dạng kiểu hình 1).
Kiểu ba alen (OCA1): Sự kết hợp ba alen của gen TYR có thể gây ra OCA nhẹ 2).
QTại sao việc biết loại bạch tạng lại quan trọng?
A
Vì tiên lượng thị lực, sự hiện diện của biến chứng toàn thân và kiểu di truyền khác nhau tùy theo loại, nên chẩn đoán chính xác phân nhóm là quan trọng. Đặc biệt HPS có kèm xu hướng chảy máu, xơ phổi và bệnh viêm ruột, cần thận trọng trước khi phẫu thuật hoặc nhổ răng. Xác định phân nhóm bằng xét nghiệm di truyền liên quan trực tiếp đến việc lập kế hoạch quản lý thích hợp.
OCA điển hình có thể được chẩn đoán lâm sàng dựa trên sự kết hợp của tóc trắng, da trắng và các dấu hiệu mắt (xuyên sáng mống mắt, rung giật nhãn cầu). Tuy nhiên, ở OA1 người Nhật, sắc tố còn tồn tại ở mống mắt, do đó chẩn đoán có thể bị chậm nếu bệnh nhân đến khám chỉ với triệu chứng mắt.
Kính hiển vi đèn khe: Xác nhận xuyên sáng mống mắt. Thực hiện đánh giá mức độ.
OCT (Chụp cắt lớp quang học): Cần thiết để phân loại thiểu sản hố trung tâm. Có thể thực hiện ở trẻ em, giúp quyết định can thiệp sớm 6).
VEP (Điện thế gợi thị giác): Quan trọng nhất để phát hiện bắt chéo bất thường của đường thị giác. Sử dụng VEP 3 chuyển đạo để đánh giá bất đối xứng sóng giữa hai bán cầu 2). Định lượng bằng hệ số giao thoa (-1 bất đối xứng tối đa, +1 bình thường) 2).
Đo góc lambda: Góc lambda > 5 độ là chỉ số lâm sàng mạnh gợi ý bạch tạng2).
Kính hiển vi điện tử tiểu cầu: Dùng để chẩn đoán xác định HPS. Thiếu hạt đặc là dấu hiệu đặc trưng 4).
Xét nghiệm di truyền: Giải trình tự toàn bộ exome (WES) 1) hoặc giải trình tự bảng gen 2, 4, 5). Đặc biệt quan trọng khi kiểu hình nhẹ hoặc không điển hình.
Không có phương pháp điều trị triệt để. Mục tiêu điều trị là tối đa hóa chức năng thị giác và quản lý biến chứng.
Quản lý Thị giác
Chỉnh khúc xạ: Đeo kính từ khi còn nhỏ là cơ bản. Can thiệp sớm để phòng ngừa nhược thị là quan trọng.
Kính chống sáng: Sử dụng thấu kính chặn khoảng 20% ánh sáng trong nhà và khoảng 80% ngoài trời 5).
Kính áp tròng có mống mắt: Có thể sử dụng để cải thiện thẩm mỹ và giảm chói sáng.
Chăm sóc thị lực kém: Sử dụng các dụng cụ hỗ trợ như kính lúp, máy đọc phóng đại và máy tính bảng.
Rung giật nhãn cầu và Lác
Phẫu thuật lác: Được thực hiện vì mục đích thẩm mỹ. Phẫu thuật Dawson-Trick-Litzkow (DTL) đôi khi được thực hiện để giảm rung giật nhãn cầu.
Điều trị rung giật nhãn cầu: Không có điều trị triệt để. Phẫu thuật lác được xem xét để xử lý tư thế đầu bất thường (đảm bảo vùng null).
Quản lý Da và Toàn thân
Bảo vệ ánh nắng: Kem chống nắng chặn tia UVB, quần áo và mũ để bảo vệ da. Nguy cơ ung thư biểu mô tế bào vảy cần được chú ý ngay cả ở người Nhật.
Quản lý HPS: NSAID (bao gồm aspirin) về nguyên tắc chống chỉ định vì làm xấu đi rối loạn chức năng tiểu cầu. Theo dõi chức năng phổi định kỳ từ tuổi thiếu niên được khuyến cáo 4).
Tư vấn di truyền: Cần thiết cho kế hoạch hóa gia đình, chẩn đoán người mang gen và xác định phân nhóm.
Đối với xơ phổi kết hợp với HPS, thuốc chống xơ hóa được sử dụng.
Liu và cộng sự (2025) báo cáo một trường hợp HPS với đột biến đồng hợp tử mới ở HPS4, trong đó xơ phổi ổn định trong 18 tháng sau khi dùng nintedanib 4). Pirfenidone cũng là một lựa chọn điều trị được sử dụng tương tự.
QLiệu pháp gen cho bệnh bạch tạng đã được ứng dụng thực tế chưa?
A
Hiện tại, liệu pháp gen cho bệnh bạch tạng chưa được ứng dụng thực tế. Liệu pháp gen đã được phê duyệt cho các bệnh võng mạc di truyền khác như bệnh tách lớp võng mạc và LCA (ví dụ: Luxturna), và nghiên cứu ứng dụng cho bệnh bạch tạng đang được tiến hành 6). Hiện tại, chỉnh khúc xạ và chăm sóc thị lực kém là trọng tâm của điều trị tiêu chuẩn.
Sự tổng hợp melanin diễn ra theo con đường sau trong melanosome:
Tyrosine → L-DOPA → DOPA quinone → (eumelanin hoặc pheomelanin)
Sự chuyển đổi dopachrome thành DHICA (axit 5,6-dihydroxyindole-2-carboxylic) được xúc tác bởi DCT (TYRP2) 2), một phản ứng thiết yếu cho tổng hợp eumelanin.
Rối loạn trưởng thành melanosome trong OCA2: Suy giảm chức năng protein OCA2 làm cản trở quá trình chuyển đổi melanosome sang giai đoạn IV (melanosome trưởng thành), làm tăng melanosome chưa trưởng thành giai đoạn I/II 1). Cơ chế chính là sự phá vỡ điều hòa pH thông qua kiểm soát dòng chloride 1).
Khi sắc tố của biểu mô sắc tố võng mạc không phù hợp, tác dụng chắn sáng trong quá trình phát triển bị mất, cản trở sự phát triển hình thái của hố trung tâm (hình thành vùng vô mạch và sự di chuyển dày đặc của tế bào hình nón). Đây là nguyên nhân gốc rễ của thị lực kém ở bệnh bạch tạng.
Ở bệnh bạch tạng, các sợi từ võng mạc thái dương bắt chéo quá mức sang bên đối diện tại giao thoa thị giác, dẫn đến nhiều sợi được xử lý như sợi mũi hơn. Sự “định hướng sai” này có thể được phát hiện dưới dạng bất đối xứng của hệ số giao thoa trên VEP2).
Trong nghiên cứu OCA8 của Rateaux và cộng sự (2025), mô hình chuột mang đột biến DCT làm giảm L-DOPA xuống 50% so với loại hoang dã đã được sử dụng, và việc bổ sung L-DOPA đã phục hồi các bất thường về mắt 2). Điều này cho thấy vai trò của L-DOPA như một chất trung gian tổng hợp melanin trong sự hình thành đường thị giác.
Đột biến GPR143 (OA1): Mất chức năng của protein GPR143 làm rối loạn vận chuyển túi melanosome, dẫn đến hình thành các melanosome lớn 3).
HPS (Rối loạn phức hợp BLOC): Mất chức năng BLOC-3 làm suy giảm hoạt hóa Rab32/38, gây ra sự phá vỡ tổng thể quá trình sinh tổng hợp các bào quan liên quan đến melanosome (LRO) 4). Các hạt đậm đặc trong tiểu cầu và thể phiến trong tế bào biểu mô phổi loại II bị ảnh hưởng. Sự tích tụ các chất giống ceroid từ các thể phiến bất thường được cho là có liên quan đến xơ phổi 4).
BLOC-1 (HPS-11): Hoạt động như một phức hợp gồm 8 tiểu đơn vị bao gồm BLOC1S5 (BLOS3), điều hòa quá trình tái chế nội bào 5).
7. Nghiên cứu mới nhất và triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
L-DOPA là chất trung gian trong tổng hợp melanin và đóng vai trò quan trọng trong sự phát triển võng mạc và hình thành đường thị giác.
Trong mô hình chuột OCA8 của Rateaux và cộng sự (2025), sự giảm L-DOPA do đột biến DCT là yếu tố chính gây ra kiểu hình mắt, và việc bổ sung L-DOPA đã cải thiện các bất thường 2). Việc ứng dụng trên người đang được nghiên cứu 6).
Việc đánh giá cải thiện chức năng thị giác bằng cách dùng levodopa ở bệnh nhân bạch tạng đang được tiến hành, với kỳ vọng về hiệu quả thúc đẩy phát triển võng mạc6).
Nitisinone, một chất ức chế 4-hydroxyphenylpyruvate dioxygenase (4-HPPD), đang được nghiên cứu về khả năng tăng melanin nội nhãn thông qua điều chỉnh con đường chuyển hóa tyrosine 6).
Nghiên cứu cơ bản về liệu pháp bổ sung gen nhắm vào các gen liên quan đến OCA (TYR, OCA2, v.v.) đang được tiến hành. Việc phê duyệt Luxturna (bổ sung gen RPE65) cho LCA (bệnh mù bẩm sinh Leber) đang thúc đẩy nghiên cứu liệu pháp gen cho các bệnh võng mạc di truyền nói chung 6).
Jiang và cộng sự (2024) đã xác định một đột biến dị hợp tử phức hợp mới (c.635A>G/c.2359+1G>T) trên gen OCA2 thông qua giải trình tự toàn bộ exome (WES) ở một gia đình người Trung Quốc 1). Phân tích chức năng xác nhận rằng protein đột biến bị suy giảm khả năng di chuyển đến tế bào hắc tố so với loại hoang dã.
Boeckelmann và cộng sự (2021) đã mô tả chi tiết phổ lâm sàng của HPS-11 do đột biến BLOC1S5 thông qua nghiên cứu 5 trường hợp bao gồm các trường hợp được báo cáo từ năm 2020 5). Các hướng dẫn theo dõi triệu chứng phổi, mắt và thần kinh đã được đưa ra.
Hiệu quả của liệu pháp chống xơ hóa với nintedanib và pirfenidone đối với xơ phổi HPS đang được đánh giá liên tục 4).
Flynn và cộng sự (2025) báo cáo rằng khi khám đáy mắt ở phụ nữ mang đột biến GPR143 mắc OA1, 94% có thay đổi sắc tố dạng mảng trước vòm và 74% có hiện tượng xuyên sáng mống mắt3). Điều này cho thấy các bất thường về mắt thường gặp ngay cả ở người mang gen.
QBệnh nhân bạch tạng có nguy cơ cao bị ung thư da không?
A
Trong OCA, do thiếu sắc tố da, tổn thương DNA do tia UV dễ tích tụ, làm tăng đáng kể nguy cơ ung thư da như ung thư biểu mô tế bào vảy. Nguy cơ được cho là cao hơn đặc biệt ở bệnh nhân OCA gốc Phi. Khuyến cáo khám da liễu định kỳ và bảo vệ da khỏi ánh nắng mặt trời nghiêm ngặt (kem chống nắng, quần áo che chắn).
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.