L’albinisme est un groupe de maladies héréditaires caractérisées par une absence ou une diminution de la pigmentation de la peau, des cheveux et des yeux, due à des mutations génétiques affectant la biosynthèse ou le transport de la mélanine. Outre la dépigmentation, ces maladies se caractérisent par des anomalies oculaires et visuelles spécifiques telles que l’hypoplasie fovéale, le nystagmus et la décussation anormale des voies visuelles.
Il n’existe pas de traitement curatif et les troubles de la fonction visuelle sont non progressifs (stationnaires). Au Japon, cette maladie est désignée comme maladie rare (loi sur les maladies rares).
Prévalence : environ 1/17 000 dans le monde. Chez les Caucasiens, l’OCA1 représente environ 50 %, tandis que chez les Africains, l’OCA2 est le plus fréquent (1/10 000)1). Chez les Chinois Han, l’OCA1 représente 70,1 % et l’OCA2 10,2 %1).
Albinisme oculaire (AO)
Hérédité récessive liée à l’X : survient chez les hommes. La pigmentation de la peau et des cheveux est normale ou légèrement réduite, les manifestations oculaires sont prédominantes.
Mutation du gène GPR143 : impliqué dans la signalisation de la biogenèse des mélanosomes. 192 mutations ont été rapportées3).
Femmes conductrices : présentent un motif en mosaïque de zones hypopigmentées au fond d’œil, semblable à des éclaboussures de boue. Environ 80 % présentent des anomalies pigmentaires rétiniennes3).
Albinisme syndromique
Syndrome de Hermansky-Pudlak (HPS) : AOC + dysfonction plaquettaire (tendance hémorragique) + trouble de la biogenèse des LRO. 11 sous-types existent4, 5). Peut se compliquer de fibrose pulmonaire et de maladies inflammatoires de l’intestin.
Syndrome de Chediak-Higashi (CHS) : AOC + immunodéficience + troubles neurologiques. Mutation du gène LYST.
12 gènes : 12 gènes ont été identifiés pour l’albinisme syndromique2).
La prévalence mondiale est d’environ 1/17 0001), et en Europe, elle est rapportée à environ 1/12 0002). Chez les Japonais, la distribution est de 34 % pour l’OCA1, 27 % pour l’OCA4 et 10 % pour le HPS1.
QL'albinisme est-il héréditaire ? Peut-il survenir même si personne dans la famille n'est atteint ?
A
L’OCA est une maladie autosomique récessive : si les deux parents sont porteurs d’un allèle muté chacun, le risque pour l’enfant de développer la maladie est de 25 %. Comme les parents peuvent être phénotypiquement normaux, l’absence d’antécédents familiaux n’est pas rare. L’OA est liée à l’X et récessive, touchant principalement les hommes, la mère étant conductrice. Les tests génétiques et le conseil génétique sont importants.
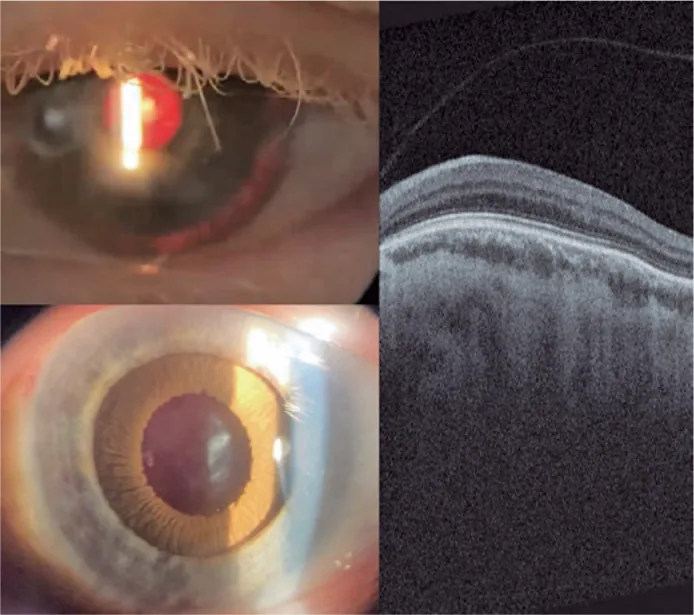
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Photographie à la lampe à fente de l’œil gauche 30 jours après la chirurgie, montrant l’iris prothétique sur le sulcus ciliaire en rétroéclairage et après mydriase pharmacologique. OCT rétinienne montrant l’absence de fossette fovéale et la persistance des couches internes de la rétine dans la zone attendue de la fovéa (hypoplasie maculaire).
Hypoplasie fovéale : facteur le plus important de la mauvaise vision. L’OCT confirme l’absence de dépression fovéale. L’angiographie à la fluorescéine montre souvent une absence de zone avasculaire périmaculaire. Dans l’OCA8, tous les cas présentent un grade 3, et le grade est corrélé à l’acuité visuelle2).
Transillumination irienne : en raison du manque de pigment irien, on observe une transillumination accrue à la lampe à fente. Dans l’OCA8, tous les cas sont de grade 32).
Nystagmus (INS) : le nystagmus à direction alternée asymétrique est observé dans environ 10 % des INS, et jusqu’à 37 % dans l’INS lié à l’albinisme2).
Mauvais décussation des voies visuelles (chiasmal misrouting) : les fibres rétiniennes temporales, qui devraient normalement suivre une voie ipsilatérale (non croisée), décussent de manière excessive vers le côté controlatéral au niveau du chiasma. Détecté par VEP. Dans l’OCA8, un coefficient chiasmatique de −0,97/−0,90 indique une asymétrie sévère2).
Strabisme : observé dans 53 à 90,5 % des cas2).
Fond d’œil hypopigmenté : le manque de pigment dans l’épithélium pigmentaire rétinien permet de voir les vaisseaux choroïdiens.
Fond d’œil des porteurs d’OA1 : 94 % présentent des modifications pigmentaires en bandes antérieures à l’arcade, et 74 % une transillumination irienne3). Des taches de dépigmentation dans la rétine périphérique (fond d’œil en mosaïque) peuvent être observées, utiles pour identifier les porteurs.
QL'acuité visuelle dans l'albinisme change-t-elle au cours de la vie ?
A
Les troubles visuels causés par l’albinisme sont stationnaires (non progressifs) et ne s’aggravent pas avec l’âge. Cependant, si une correction réfractive appropriée ou un traitement de l’amblyopie n’est pas effectué, l’acuité visuelle peut rester fixe sans amélioration. Une intervention précoce par correction réfractive et soins de basse vision est importante.
Les principaux gènes responsables et les caractéristiques de chaque sous-type sont présentés ci-dessous.
Sous-type
Gène responsable
Principales caractéristiques
OCA1
TYR
Déficit en tyrosinase
OCA2
OCA2 (gène P)
Régulation du pH des mélanosomes1)
OCA3
TYRP1
Synthèse de l’eumélanine
OCA4
SLC45A2
Transporteur mélanosomique
OCA8
DCT (TYRP2)
Conversion de la dopachrome2)
OA1
GPR143
Signal mélanosomique3)
HPS1/HPS4
HPS1/HPS4
Complexe BLOC-34)
HPS11
BLOC1S5
Complexe BLOC-15)
Fonction de la protéine OCA2 : Elle appartient à la famille des antiporteurs Na⁺/H⁺ avec une structure à 12 hélices α transmembranaires 1). En tant que composant d’un canal anionique spécifique au mélanosome, elle régule le pH des mélanosomes de stade I/II via le contrôle du courant chlorure 1). ClinVar répertorie 477 mutations pathogènes 1).
DCT (TYRP2) : Catalyse la conversion de la dopachrome en DHICA (acide 5,6-dihydroxyindole-2-carboxylique) 2). C’est le gène responsable de l’OCA8.
GPR143 (OA1) : Molécule de signalisation régulant le transport vésiculaire des mélanosomes. Les mutations entraînent la formation de macromélanosomes 3).
HPS (complexe BLOC) : HPS1/HPS4 fonctionnent comme un complexe BLOC-3 agissant comme GEF (facteur d’échange de nucléotides guanyliques) pour Rab32/38 4). BLOC-1 est composé de 8 sous-unités et participe au recyclage endosomal ; la mutation du gène BLOC1S5 (BLOS3) provoque le HPS-11 (décrit pour la première fois en 2020) 5).
Consanguinité : La prévalence augmente dans les populations où les mariages consanguins sont fréquents, comme au Pakistan 4).
Différences raciales et régionales : La fréquence est élevée dans certaines régions d’Afrique.
Mutations hétérozygotes composites : Dans l’OCA2, de nouvelles mutations hétérozygotes composites ont été identifiées, contribuant à la diversité phénotypique 1).
Type triallélique (OCA1) : Des combinaisons de trois allèles du gène TYR peuvent entraîner une OCA légère 2).
QPourquoi est-il important de connaître le type d'albinisme ?
A
Le pronostic visuel, la présence de complications systémiques et le mode de transmission diffèrent selon le type, il est donc important de poser un diagnostic précis du sous-type. En particulier, le syndrome de Hermansky-Pudlak (HPS) s’accompagne d’une tendance aux saignements, d’une fibrose pulmonaire et d’une maladie inflammatoire de l’intestin, nécessitant des précautions avant une intervention chirurgicale ou une extraction dentaire. L’identification du sous-type par test génétique est directement liée à l’élaboration d’un plan de prise en charge approprié.
L’albinisme oculo-cutané (OCA) typique peut être diagnostiqué cliniquement par la combinaison de cheveux blancs, de peau blanche et de signes oculaires (transillumination de l’iris, nystagmus). Cependant, dans l’albinisme oculaire de type 1 (OA1) chez les Japonais, le pigment persiste dans l’iris, donc le diagnostic peut être retardé si le patient consulte uniquement pour des symptômes oculaires.
Lampe à fente : Confirmation de la transillumination de l’iris. Évaluation du grade.
OCT (tomographie par cohérence optique) : Essentiel pour le grading de l’hypoplasie fovéolaire. Réalisable même chez l’enfant, utile pour décider d’une intervention précoce6).
VEP (potentiels évoqués visuels) : Le plus important pour détecter une décussation anormale des voies visuelles. Évaluation de l’asymétrie des ondes entre les hémisphères gauche et droit à l’aide d’un VEP à 3 dérivations2). Quantification par le coefficient de décussation ( −1 : asymétrie maximale, +1 : normal)2).
Mesure de l’angle lambda : Un angle lambda > 5 degrés est un indicateur clinique fort d’albinisme2).
Microscopie électronique des plaquettes : Utilisée pour le diagnostic définitif du HPS. L’absence de granules denses est une caractéristique4).
Test génétique : Séquençage de l’exome entier (WES)1) ou séquençage par panel2, 4, 5). Particulièrement important en cas de phénotype léger ou atypique.
Il n’existe pas de traitement curatif. Le traitement vise à maximiser la fonction visuelle et à gérer les complications.
Prise en charge visuelle
Correction de la réfraction : Le port de lunettes dès la petite enfance est la base. Une intervention précoce pour prévenir l’amblyopie est importante.
Lunettes filtrantes : Utiliser des verres filtrant environ 20 % de la lumière à l’intérieur et environ 80 % à l’extérieur 5).
Lentilles de contact à iris : Peuvent être utilisées pour améliorer l’apparence et réduire la photophobie.
Soins basse vision : Utiliser des aides comme les loupes, les loupes électroniques et les tablettes.
Nystagmus et strabisme
Chirurgie du strabisme : Réalisée à des fins esthétiques. Parfois, l’opération de Dawson-Trick-Litzkow (DTL) est pratiquée pour réduire le nystagmus.
Traitement du nystagmus : Il n’existe pas de traitement curatif. La chirurgie du strabisme peut être envisagée pour corriger une position anormale de la tête (zone nulle).
Gestion cutanée et générale
Protection solaire : Protection cutanée par écran solaire bloquant les UVB, vêtements et chapeaux. Même chez les Japonais, il faut être attentif au risque de carcinome épidermoïde.
Gestion du HPS : Les AINS (y compris l’aspirine) sont généralement contre-indiqués car ils aggravent le dysfonctionnement plaquettaire. Une surveillance régulière de la fonction pulmonaire à partir de l’adolescence est recommandée 4).
Conseil génétique : Essentiel pour la planification familiale, le diagnostic de porteur et la détermination du sous-type.
Traitement médicamenteux de la fibrose pulmonaire du HPS
Pour la fibrose pulmonaire associée au HPS, des médicaments antifibrosants sont utilisés.
Liu et al. (2025) ont rapporté un cas de HPS avec une nouvelle mutation homozygote de HPS4, où l’administration de nintédanib a stabilisé la fibrose pulmonaire pendant 18 mois 4). La pirfénidone est également une option thérapeutique utilisée de manière similaire.
QLa thérapie génique pour l'albinisme est-elle disponible en pratique clinique ?
A
À ce jour, la thérapie génique pour l’albinisme n’est pas encore disponible en pratique clinique. Pour d’autres maladies rétiniennes héréditaires comme la rétinoschisis et l’ACL, des thérapies géniques ont été approuvées (ex. Luxturna), et des recherches sur leur application à l’albinisme sont en cours6). Actuellement, la correction de la réfraction et les soins de basse vision constituent le traitement standard.
La synthèse de la mélanine se déroule dans les mélanosomes selon la voie suivante :
Tyrosine → L-DOPA → DOPAquinone → (eumélanine ou phéomélanine)
La conversion de la dopachrome en DHICA (acide 5,6-dihydroxyindole-2-carboxylique) est catalysée par la DCT (TYRP2)2), une réaction essentielle pour la synthèse de l’eumélanine.
Trouble de la maturation des mélanosomes dans l’OCA2 : La diminution de la fonction de la protéine OCA2 entraîne un blocage de la transition des mélanosomes vers le stade IV (mélanosomes matures), avec une augmentation des mélanosomes immatures aux stades I/II1). Le mécanisme principal est une perturbation de la régulation du pH par le contrôle du courant chlorure1).
Lorsque la pigmentation de l’épithélium pigmentaire rétinien est inadéquate, l’effet de protection contre la lumière pendant le développement est perdu, ce qui entrave le développement morphologique de la fovéa (formation de la zone avasculaire et migration dense des cônes). C’est la cause fondamentale de la mauvaise acuité visuelle dans l’albinisme.
Mécanisme de la décussation anormale des voies visuelles
Dans l’albinisme, les fibres provenant de la rétine temporale décussent de manière excessive vers le côté controlatéral au niveau du chiasma, de sorte qu’un plus grand nombre de fibres sont traitées comme nasales. Ce « misrouting » est détecté par les VEP comme une asymétrie du coefficient chiasmatique2).
Dans leur étude OCA8 de 2025, Rateaux et al. ont utilisé un modèle murin présentant une mutation DCT réduisant la L-DOPA à 50 % du taux sauvage, et ont montré que la supplémentation en L-DOPA corrigeait les anomalies ophtalmiques 2). Cela suggère que la L-DOPA, en tant qu’intermédiaire de la synthèse de la mélanine, joue un rôle dans la formation des voies visuelles.
Mutation GPR143 (OA1) : La perte de fonction de la protéine GPR143 perturbe le transport vésiculaire des mélanosomes, conduisant à la formation de macromélanosomes 3).
HPS (troubles du complexe BLOC) : La perte de fonction de BLOC-3 altère l’activation de Rab32/38, entraînant une perturbation générale de la biogenèse des organites associés aux mélanosomes (LRO : lysosome-related organelle) 4). Les corps denses des plaquettes et les corps lamellaires des cellules épithéliales pulmonaires de type II sont affectés. L’accumulation de substances céroïdes issues de corps lamellaires anormaux serait impliquée dans la fibrose pulmonaire 4).
BLOC-1 (HPS-11) : Fonctionne comme un complexe de 8 sous-unités incluant BLOC1S5 (BLOS3) et régule le recyclage endosomal 5).
7. Recherches récentes et perspectives futures (études en phase de recherche)
La L-DOPA est un intermédiaire de la synthèse de la mélanine et joue un rôle important dans le développement rétinien et la formation des voies visuelles.
Dans le modèle murin OCA8 de Rateaux et al. (2025), la diminution de L-DOPA due à la mutation DCT était le principal facteur du phénotype ophtalmique, et la supplémentation en L-DOPA a amélioré les anomalies 2). Des applications chez l’humain sont à l’étude 6).
Une évaluation de l’amélioration de la fonction visuelle par administration de lévodopa chez des patients atteints d’albinisme est en cours, avec un effet promoteur attendu sur le développement rétinien 6).
La nitisinone, un inhibiteur de la 4-hydroxyphénylpyruvate dioxygénase (4-HPPD), est étudiée pour son potentiel à augmenter la mélanine intraoculaire en régulant la voie métabolique de la tyrosine 6).
La recherche fondamentale sur la thérapie génique de substitution ciblant les gènes associés à l’OCA (TYR, OCA2, etc.) progresse. L’approbation de Luxturna (substitution du gène RPE65) pour la LCA (amaurose congénitale de Leber) accélère la recherche en thérapie génique pour les maladies rétiniennes héréditaires en général 6).
Jiang et al. (2024) ont identifié une nouvelle mutation composite hétérozygote (c.635A>G/c.2359+1G>T) du gène OCA2 par séquençage complet de l’exome (WES) dans une famille chinoise 1). L’analyse fonctionnelle a confirmé que la protéine mutante présente un défaut de translocation vers les mélanocytes par rapport au type sauvage.
Boeckelmann et al. (2021) ont détaillé le spectre clinique du HPS-11 dû à une mutation de BLOC1S5 dans une étude de 5 cas, incluant des cas rapportés depuis 2020 5). Des directives de surveillance des symptômes pulmonaires, oculaires et neurologiques sont fournies.
L’efficacité du traitement antifibrotique par nintedanib et pirfénidone pour la fibrose pulmonaire du HPS est en cours d’évaluation continue 4).
Flynn et al. (2025) ont rapporté que chez les femmes porteuses d’OA1 avec mutation GPR143, l’examen du fond d’œil a révélé des modifications pigmentaires maculaires en arcade antérieure chez 94 % et une transillumination irienne chez 74 % 3). Il a été démontré que les anomalies ophtalmiques sont fréquentes même chez les porteurs.
QLes patients atteints d'albinisme ont-ils un risque élevé de cancer de la peau ?
A
Dans l’OCA, l’absence de pigment cutané entraîne une accumulation de dommages à l’ADN causés par les UV, augmentant significativement le risque de cancers cutanés tels que le carcinome épidermoïde. Le risque est particulièrement élevé chez les patients OCA d’origine africaine. Des consultations dermatologiques régulières et une protection solaire stricte (écran solaire, vêtements couvrants) sont recommandées.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.