L’albinismo è un gruppo di malattie ereditarie caratterizzate da assenza o riduzione della pigmentazione della pelle, dei capelli e degli occhi, dovuta a mutazioni genetiche che interessano la biosintesi o il trasporto della melanina. Oltre alla mancanza di pigmento, questo gruppo di malattie è caratterizzato da specifiche anomalie oculari e delle vie visive, come ipoplasia foveale, nistagmo e decussazione anomala delle vie visive.
Non esiste una terapia curativa e il deficit visivo è non progressivo (stazionario). In Giappone questa malattia è designata come malattia rara (legge sulle malattie rare).
Prevalenza: circa 1/17.000 nel mondo. Nei caucasici, l’AOC1 rappresenta circa il 50%, mentre negli africani l’AOC2 è il più comune (1/10.000)1). Negli Han cinesi, l’AOC1 è il 70,1% e l’AOC2 il 10,2%1).
Albinismo oculare (AO)
Ereditarietà recessiva legata all’X: si manifesta nei maschi. La pigmentazione di pelle e capelli è normale o lievemente ridotta, predominano i segni oculari.
Mutazione del gene GPR143: coinvolto nella trasduzione del segnale della biogenesi dei melanosomi. Sono state riportate 192 mutazioni3).
Donne portatrici: mostrano un pattern a mosaico di ipopigmentazione a chiazze al fondo dell’occhio. Circa l’80% presenta anomalie pigmentarie retiniche3).
Albinismo sindromico
Sindrome di Hermansky-Pudlak (HPS): AOC + disfunzione piastrinica (tendenza al sanguinamento) + alterazione della biogenesi degli LRO. Esistono 11 sottotipi4, 5). Può complicarsi con fibrosi polmonare e malattie infiammatorie intestinali.
Sindrome di Chediak-Higashi (CHS): AOC + immunodeficienza + disturbi neurologici. Mutazione del gene LYST.
12 geni: per l’albinismo sindromico sono stati identificati 12 geni2).
La prevalenza mondiale è di circa 1/17.0001), mentre in Europa è riportata circa 1/12.0002). Nei giapponesi è nota una distribuzione del 34% per AOC1, 27% per AOC4 e 10% per HPS1.
QL'albinismo è ereditario? Può manifestarsi anche se in famiglia non ci sono persone con albinismo?
A
L’OCA è una malattia autosomica recessiva: se entrambi i genitori sono portatori di un allele mutato ciascuno, il rischio per il figlio di sviluppare la malattia è del 25%. Poiché i genitori possono essere fenotipicamente normali, l’assenza di storia familiare non è rara. L’OA è recessiva legata all’X, colpisce principalmente i maschi, la madre è portatrice. I test genetici e la consulenza genetica sono importanti.
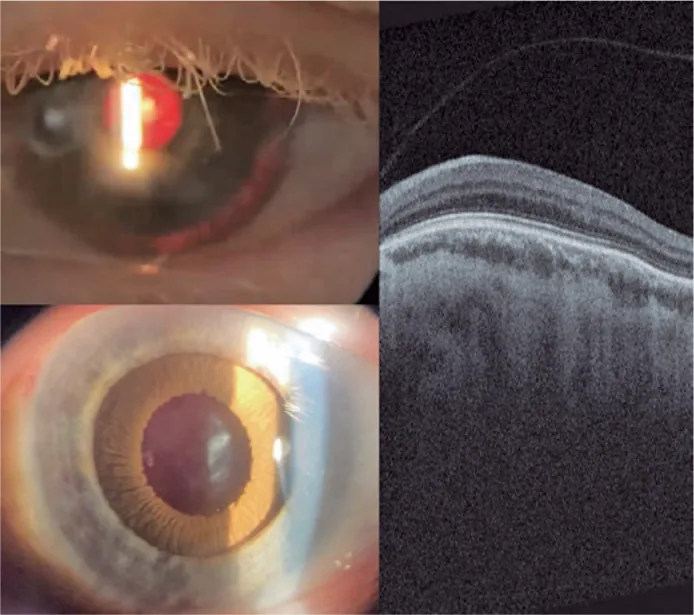
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Fotografia con lampada a fessura dell’occhio sinistro 30 giorni dopo l’intervento, che mostra l’iride protesica sul solco ciliare in retroilluminazione e dopo midriasi farmacologica. OCT retinico che mostra assenza della fossetta foveale e persistenza degli strati interni della retina nell’area prevista della fovea (ipoplasia maculare).
Ipoplasia foveale: fattore più importante per la scarsa visione. L’OCT conferma l’assenza della depressione foveale. All’angiografia con fluoresceina spesso manca la zona avascolare perimaculare. Nell’OCA8 tutti i casi presentano grado 3, e il grado è correlato all’acuità visiva2).
Transilluminazione iridea: a causa della mancanza di pigmento irideo, alla lampada a fessura si osserva un aumento della transilluminazione. Nell’OCA8 tutti i casi sono di grado 32).
Nistagmo (INS): il nistagmo asimmetrico periodico alternante si osserva in circa il 10% di tutti gli INS, e fino al 37% nell’INS da albinismo2).
Decussazione anomala delle vie ottiche (chiasmal misrouting): le fibre retiniche temporali, che normalmente seguono un percorso ipsilaterale (non incrociato), decussano eccessivamente verso il lato controlaterale al chiasma. Rilevato con VEP. Nell’OCA8 è stato riportato un coefficiente chiasmatico di −0,97/−0,90, indicante una grave asimmetria2).
Strabismo: presente nel 53-90,5% dei casi2).
Fondo ipopigmentato: la mancanza di pigmento nell’epitelio pigmentato retinico permette di vedere i vasi coroideali.
Fondo dei portatori di OA1: il 94% presenta alterazioni pigmentarie a bande anteriormente all’arcata, il 74% transilluminazione iridea3). Nella retina periferica possono essere presenti chiazze depigmentate (fondo a mosaico), utili per identificare i portatori.
QL'acuità visiva nell'albinismo cambia nel corso della vita?
A
I disturbi visivi causati dall’albinismo sono stazionari (non progressivi) e non peggiorano con l’età. Tuttavia, se non vengono effettuate una corretta correzione refrattiva o una terapia per l’ambliopia, l’acuità visiva può rimanere fissa senza miglioramento. È importante un intervento precoce con correzione refrattiva e cure per la bassa visione.
I principali geni responsabili e le caratteristiche di ciascun sottotipo sono mostrati di seguito.
Sottotipo
Gene responsabile
Caratteristiche principali
OCA1
TYR
Deficit di tirosinasi
OCA2
OCA2 (gene P)
Regolazione del pH dei melanosomi1)
OCA3
TYRP1
Sintesi di eumelanina
OCA4
SLC45A2
Trasportatore melanosomiale
OCA8
DCT (TYRP2)
Conversione della dopacromo2)
OA1
GPR143
Segnale melanosomiale3)
HPS1/HPS4
HPS1/HPS4
Complesso BLOC-34)
HPS11
BLOC1S5
Complesso BLOC-15)
Funzione della proteina OCA2 : Appartiene alla famiglia degli antiporti Na⁺/H⁺ con una struttura a 12 eliche α transmembrana 1). Come componente di un canale anionico specifico del melanosoma, regola il pH dei melanosomi di stadio I/II attraverso il controllo della corrente di cloruro 1). ClinVar elenca 477 mutazioni patogene 1).
DCT (TYRP2) : Catalizza la conversione della dopacromo in DHICA (acido 5,6-diidrossiindolo-2-carbossilico) 2). È il gene responsabile dell’OCA8.
GPR143 (OA1) : Molecola di segnalazione che regola il trasporto vescicolare dei melanosomi. Le mutazioni portano alla formazione di macromelanosomi 3).
HPS (complesso BLOC) : HPS1/HPS4 funzionano come complesso BLOC-3 agendo come GEF (fattore di scambio della guanina nucleotide) per Rab32/38 4). BLOC-1 è composto da 8 subunità ed è coinvolto nel riciclo endosomiale; la mutazione del gene BLOC1S5 (BLOS3) causa HPS-11 (descritto per la prima volta nel 2020) 5).
Consanguineità : Nelle popolazioni con alta frequenza di matrimoni consanguinei, come in Pakistan, la prevalenza aumenta 4).
Differenze etniche e regionali : In alcune regioni dell’Africa la frequenza è elevata.
Mutazioni eterozigoti composte : Nell’OCA2 sono state identificate nuove mutazioni eterozigoti composte, che contribuiscono alla diversità fenotipica 1).
Tipo triallelico (OCA1) : Combinazioni di tre alleli del gene TYR possono causare OCA lieve 2).
QPerché è importante conoscere il tipo di albinismo?
A
La prognosi visiva, la presenza di complicanze sistemiche e la modalità di trasmissione differiscono a seconda del tipo, pertanto è importante una diagnosi accurata del sottotipo. In particolare, la sindrome di Hermansky-Pudlak (HPS) si associa a tendenza al sanguinamento, fibrosi polmonare e malattia infiammatoria intestinale, richiedendo cautela prima di interventi chirurgici o estrazioni dentarie. L’identificazione del sottotipo tramite test genetico è direttamente collegata all’elaborazione di un piano di gestione appropriato.
L’OCA tipico può essere diagnosticato clinicamente dalla combinazione di capelli bianchi, pelle bianca e segni oculari (transilluminazione dell’iride, nistagmo). Tuttavia, nell’OA1 giapponese, il pigmento persiste nell’iride, quindi la diagnosi può essere ritardata se il paziente si presenta solo con sintomi oculari.
VEP (potenziali evocati visivi) : Il più importante per rilevare una decussazione anomala delle vie visive. Valutazione dell’asimmetria delle onde tra emisfero sinistro e destro mediante VEP a 3 derivazioni2). Quantificazione tramite coefficiente di decussazione ( −1 asimmetria massima, +1 normale)2).
Misurazione dell’angolo lambda : Un angolo lambda > 5 gradi è un forte indicatore clinico di albinismo2).
Angiografia retinica con fluoresceina (FA) : Chiarisce il pattern a mosaico del fondo oculare nei portatori di OA1.
Microscopia elettronica delle piastrine : Utilizzata per la diagnosi definitiva di HPS. L’assenza di granuli densi è un reperto caratteristico4).
Test genetico : Sequenziamento dell’intero esoma (WES)1) o sequenziamento di pannelli2, 4, 5). Particolarmente importante in caso di fenotipo lieve o atipico.
Non esiste una terapia curativa. Il trattamento mira a massimizzare la funzione visiva e a gestire le complicanze.
Gestione visiva
Correzione refrattiva : L’uso di occhiali fin dalla prima infanzia è la base. Un intervento precoce per prevenire l’ambliopia è importante.
Occhiali schermanti : Utilizzare lenti che filtrano circa il 20% della luce in interni e circa l’80% all’aperto 5).
Lenti a contatto con iride : Possono essere utilizzate per migliorare l’aspetto estetico e ridurre la fotofobia.
Cura dell’ipovisione : Utilizzare ausili come lenti d’ingrandimento, ingranditori per lettura e tablet.
Nistagmo e strabismo
Chirurgia dello strabismo : Eseguita per scopi estetici. Talvolta viene eseguita l’operazione di Dawson-Trick-Litzkow (DTL) per ridurre il nistagmo.
Trattamento del nistagmo : Non esiste una terapia curativa. La chirurgia dello strabismo può essere presa in considerazione per correggere una posizione anomala della testa (zona nulla).
Gestione cutanea e sistemica
Protezione solare : Protezione della pelle con crema solare che blocca i raggi UVB, indumenti e cappelli. Anche nei giapponesi, prestare attenzione al rischio di carcinoma a cellule squamose.
Gestione dell’HPS : I FANS (inclusa l’aspirina) sono generalmente controindicati perché peggiorano la disfunzione piastrinica. Si raccomanda un monitoraggio regolare della funzionalità polmonare dall’adolescenza 4).
Consulenza genetica : Essenziale per la pianificazione familiare, la diagnosi di portatore e la determinazione del sottotipo.
Terapia farmacologica per la fibrosi polmonare da HPS
Per la fibrosi polmonare associata all’HPS vengono utilizzati farmaci antifibrotici.
Liu et al. (2025) hanno riportato un caso di HPS con una nuova mutazione omozigote di HPS4, in cui la somministrazione di nintedanib ha stabilizzato la fibrosi polmonare per 18 mesi 4). Anche il pirfenidone è un’opzione terapeutica utilizzata.
QLa terapia genica per l'albinismo è disponibile nella pratica clinica?
A
Ad oggi, la terapia genica per l’albinismo non è ancora stata introdotta nella pratica clinica. Per altre malattie retiniche ereditarie come la retinoschisi e l’LCA, sono state approvate terapie geniche (es. Luxturna), e la ricerca sulla loro applicazione all’albinismo è in corso6). Attualmente, la correzione refrattiva e la riabilitazione visiva sono il cardine del trattamento standard.
La sintesi della melanina avviene all’interno dei melanosomi attraverso la seguente via:
Tirosina → L-DOPA → DOPA-chinone → (eumelanina o feomelanina)
La conversione del dopacromo in DHICA (acido 5,6-diidrossiindolo-2-carbossilico) è catalizzata dalla DCT (TYRP2)2), una reazione essenziale per la sintesi dell’eumelanina.
Alterazione della maturazione dei melanosomi nell’OCA2 : La ridotta funzionalità della proteina OCA2 impedisce la transizione dei melanosomi allo stadio IV (melanosomi maturi), con un aumento dei melanosomi immaturi agli stadi I/II1). Il meccanismo principale è un’alterazione della regolazione del pH attraverso il controllo del flusso di cloro1).
Quando la pigmentazione dell’epitelio pigmentato retinico è inadeguata, l’effetto di schermatura della luce durante lo sviluppo viene perso, ostacolando lo sviluppo morfologico della fovea (formazione della zona avascolare e migrazione densa dei coni). Questa è la causa fondamentale della scarsa acuità visiva nell’albinismo.
Nell’albinismo, le fibre provenienti dalla retina temporale decussano eccessivamente verso il lato controlaterale al chiasma, così che un numero maggiore di fibre viene elaborato come nasale. Questo «misrouting» viene rilevato ai VEP come asimmetria del coefficiente chiasmatico2).
Nello studio OCA8 del 2025, Rateaux et al. hanno utilizzato un modello murino con mutazione DCT che riduce la L-DOPA al 50% del tipo selvatico, dimostrando che la supplementazione di L-DOPA corregge le anomalie oftalmiche 2). Ciò suggerisce che la L-DOPA, come intermedio della sintesi della melanina, svolge un ruolo nella formazione delle vie visive.
Mutazione GPR143 (OA1) : La perdita di funzione della proteina GPR143 compromette il trasporto vescicolare dei melanosomi, portando alla formazione di macromelanosomi 3).
HPS (disturbi del complesso BLOC) : La perdita di funzione di BLOC-3 altera l’attivazione di Rab32/38, causando un’alterazione generale della biogenesi degli organelli associati ai melanosomi (LRO: organelli correlati ai lisosomi) 4). I corpi densi delle piastrine e i corpi lamellari delle cellule epiteliali polmonari di tipo II sono colpiti. Alla base della fibrosi polmonare vi sarebbe l’accumulo di sostanze ceroidi-simili da corpi lamellari anomali 4).
BLOC-1 (HPS-11) : Funziona come un complesso di 8 subunità, inclusa BLOC1S5 (BLOS3), e regola il riciclo endosomiale 5).
7. Ricerche recenti e prospettive future (stadi di ricerca)
La L-DOPA è un intermedio della sintesi della melanina e svolge un ruolo importante nello sviluppo retinico e nella formazione delle vie visive.
Nel modello murino OCA8 di Rateaux et al. (2025), la riduzione di L-DOPA dovuta alla mutazione DCT era il fattore principale del fenotipo oftalmico, e la supplementazione di L-DOPA ha migliorato le anomalie 2). Sono in corso studi per l’applicazione nell’uomo 6).
È in corso la valutazione del miglioramento della funzione visiva mediante somministrazione di levodopa in pazienti affetti da albinismo, con un effetto promotore atteso sullo sviluppo retinico 6).
Il nitisinone, un inibitore della 4-idrossifenilpiruvato diossigenasi (4-HPPD), è studiato per il suo potenziale di aumentare la melanina intraoculare regolando la via metabolica della tirosina 6).
La ricerca di base sulla terapia genica sostitutiva mirata ai geni associati all’OCA (TYR, OCA2, ecc.) sta progredendo. L’approvazione di Luxturna (sostituzione del gene RPE65) per LCA (amaurosi congenita di Leber) sta accelerando la ricerca sulla terapia genica per le malattie retiniche ereditarie in generale 6).
Jiang et al. (2024) hanno identificato una nuova mutazione eterozigote composta (c.635A>G/c.2359+1G>T) del gene OCA2 mediante sequenziamento dell’intero esoma (WES) in una famiglia cinese 1). L’analisi funzionale ha confermato che la proteina mutante presenta un difetto di traslocazione nei melanociti rispetto al tipo selvatico.
Boeckelmann et al. (2021) hanno descritto in dettaglio lo spettro clinico dell’HPS-11 dovuto a mutazione di BLOC1S5 in uno studio di 5 casi, inclusi casi riportati dopo il 2020 5). Vengono fornite linee guida per il monitoraggio dei sintomi polmonari, oculari e neurologici.
L’efficacia della terapia antifibrotica con nintedanib e pirfenidone per la fibrosi polmonare da HPS è in continua valutazione 4).
Screening del fondo oculare nelle portatrici di OA1
Flynn et al. (2025) hanno riportato che all’esame del fondo oculare di donne portatrici di OA1 con mutazione GPR143, nel 94% sono state osservate alterazioni pigmentarie maculari ad arco anteriore e nel 74% trasilluminazione dell’iride3). È stato dimostrato che anche le portatrici presentano frequentemente anomalie oftalmologiche.
QI pazienti affetti da albinismo hanno un rischio elevato di cancro della pelle?
A
Nell’OCA, la mancanza di pigmento cutaneo facilita l’accumulo di danni al DNA indotti dai raggi UV, aumentando significativamente il rischio di tumori cutanei come il carcinoma squamocellulare. Il rischio è considerato particolarmente elevato nei pazienti OCA di origine africana. Si raccomandano visite dermatologiche regolari e una rigorosa protezione solare (crema solare, indumenti coprenti).
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.