Albinism is a group of hereditary disorders caused by genetic mutations in the biosynthesis or transport of melanin pigment, resulting in a lack or reduction of pigment in the skin, hair, and eyes. In addition to pigment deficiency, this group of disorders is characterized by specific ocular and visual pathway abnormalities such as foveal hypoplasia, nystagmus, and abnormal optic pathway decussation.
There is no curative treatment, and visual impairment is non-progressive (static). In Japan, it is designated as an intractable disease under the Intractable Disease Act.
Prevalence: Approximately 1 in 17,000 worldwide. In Caucasians, OCA1 accounts for about 50%; in Africans, OCA2 is most common (1 in 10,000)1). In Han Chinese, OCA1 is 70.1% and OCA2 is 10.2%1).
Ocular Albinism (OA)
X-linked recessive inheritance: Affects males. Skin and hair pigmentation is normal or only mildly reduced, with ocular findings being predominant.
GPR143 gene mutation: Involved in melanosome biogenesis signaling. 192 mutations have been reported3).
Female carriers: Show a mud-splatter-like mosaic hypopigmentation pattern in the fundus. Approximately 80% have retinal pigment abnormalities3).
Syndromic Albinism
Hermansky-Pudlak syndrome (HPS): OCA + platelet dysfunction (bleeding tendency) + LRO biogenesis disorder. There are 11 subtypes4, 5). May be complicated by pulmonary fibrosis and inflammatory bowel disease.
12 genes: 12 genes have been identified for syndromic albinism2).
The worldwide prevalence is approximately 1 in 17,0001), and in Europe it is reported to be about 1 in 12,0002). In Japanese individuals, the distribution is known to be OCA1 34%, OCA4 27%, and HPS1 10%.
QIs albinism inherited? Can it occur even if no one in the family has albinism?
A
OCA is an autosomal recessive disorder; if both parents are carriers with one mutant allele each, the child has a 25% chance of being affected. Since it can occur even if both parents are phenotypically normal, it is not uncommon even without a family history. OA is X-linked recessive, mainly affecting males, with the mother being a carrier. Genetic testing and genetic counseling are important.
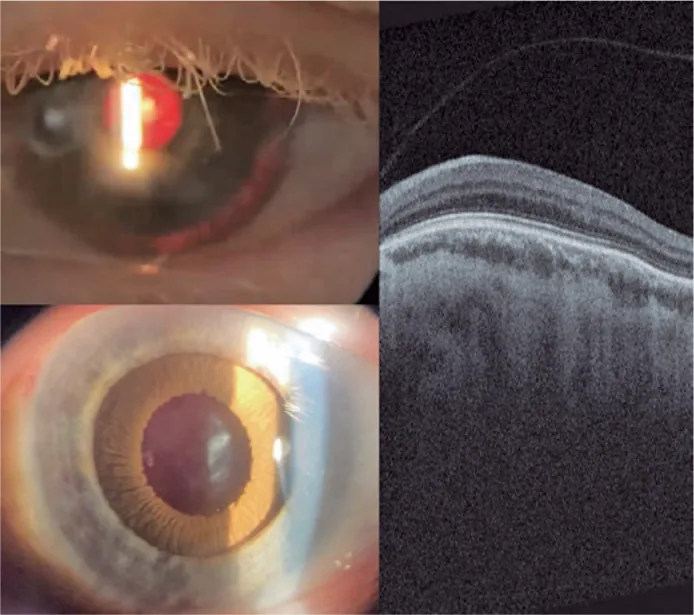
Guilherme Vieira Peixoto; Gabriela Tomaz Martinho; Caio Cezar Toledo de Conti; Eduardo Villaça Filho; Renato Klingelfus Pinheiro. Cataract surgery and artificial iris implantation in patient with oculocutaneous albinism: a case report. Arq Bras Oftalmol.; 87(4):e2022-0286 Figure 4. PMCID: PMC11620172. License: CC BY.
Left-eye slit lamp photograph 30 days after surgery, showing prosthetic iris on ciliary sulcus through retro illumination and after pharmacologic mydriasis. Retinal OCT showing absence of foveal pit and persistence of retinal inner layers through expected area of fovea (macular hypoplasia).
Foveal hypoplasia: The most important factor for poor vision. OCT confirms the absence of foveal depression. In fluorescein angiography, the absence of an avascular zone around the macula is also characteristic. In OCA8, all cases show grade 3, and the grade correlates with visual acuity2).
Iris transillumination: Due to lack of iris pigment, increased transillumination is observed on slit-lamp examination. In OCA8, all cases show grade 32).
Nystagmus (INS): Asymmetric periodic alternating nystagmus is seen in about 10% of all INS, and up to 37% in albinism INS2).
Chiasmal misrouting: Temporal retinal fibers that should normally follow an ipsilateral (uncrossed) pathway cross excessively to the contralateral side at the optic chiasm. Detected by VEP. In OCA8, chiasmal coefficients of -0.97/-0.90 indicate severe asymmetry2).
Strabismus: Observed in 53-90.5% of cases2).
Hypopigmented fundus: Choroidal vessels are visible due to lack of pigment in the retinal pigment epithelium.
Fundus of OA1 carriers: 94% show patchy pigment changes anterior to the arcuate vessels, and 74% show iris transillumination3). Depigmented spots (mosaic fundus) may be seen in the peripheral retina, useful for identifying carriers.
QDoes visual acuity in albinism change throughout life?
A
Visual dysfunction due to albinism is stationary (non-progressive) and does not worsen with age. However, if appropriate refractive correction and amblyopia treatment are not provided, visual acuity may remain fixed without improvement. Early intervention with refractive correction and low vision care is important.
The main causative genes and characteristics of each subtype are shown below.
Subtype
Causative Gene
Main Features
OCA1
TYR
Tyrosinase deficiency
OCA2
OCA2 (P gene)
Melanosome pH regulation1)
OCA3
TYRP1
Eumelanin synthesis
OCA4
SLC45A2
Melanosome transporter
OCA8
DCT (TYRP2)
Dopachrome conversion2)
OA1
GPR143
Melanosome signaling3)
HPS1/HPS4
HPS1/HPS4
BLOC-3 complex4)
HPS11
BLOC1S5
BLOC-1 complex5)
Function of OCA2 protein: Belongs to the Na⁺/H⁺ antiporter family with a 12-transmembrane α-helix structure 1). As a component of the melanosome-specific anion channel, it regulates the pH of stage I/II melanosomes through chloride current control 1). ClinVar lists 477 pathogenic mutations 1).
DCT (TYRP2): Catalyzes the conversion of dopachrome to DHICA (5,6-dihydroxyindole-2-carboxylic acid) 2). It is the causative gene for OCA8.
GPR143 (OA1): A signaling molecule that regulates melanosome vesicle transport. Mutations lead to the formation of macromelanosomes 3).
HPS (BLOC complex): HPS1/HPS4 function as a BLOC-3 complex acting as a GEF (guanine nucleotide exchange factor) for Rab32/38 4). BLOC-1 consists of 8 subunits and is involved in endosomal recycling; BLOC1S5 (BLOS3) gene mutations cause HPS-11 (first described in 2020) 5).
Consanguineous marriage: Prevalence increases in populations with high rates of consanguinity, such as in Pakistan 4).
Racial/regional differences: Frequency is high in some parts of Africa.
Compound heterozygous mutations: In OCA2, novel compound heterozygous mutations have been identified and contribute to phenotypic diversity 1).
Tri-allelic (OCA1): Combinations of three TYR gene alleles can result in mild OCA 2).
QWhy is it meaningful to know the type of albinism?
A
Since visual prognosis, presence of systemic complications, and inheritance pattern differ by type, accurate subtype diagnosis is important. In particular, HPS is associated with bleeding tendency, pulmonary fibrosis, and inflammatory bowel disease, requiring caution before surgery or tooth extraction. Subtype identification by genetic testing directly leads to appropriate management planning.
Typical OCA can be clinically diagnosed from the combination of white hair, white skin, and ocular findings (iris transillumination, nystagmus). However, in Japanese OA1, pigment remains in the iris, so diagnosis may be delayed if patients present only with ocular symptoms.
VEP (Visual Evoked Potential): Most important for detecting abnormal optic pathway decussation. Three-lead VEP is used to evaluate waveform asymmetry between the left and right hemispheres 2). Quantified by the chiasmal coefficient (−1 indicates maximum asymmetry, +1 normal) 2).
Angle lambda measurement: Angle lambda >5 degrees is a strong clinical indicator of albinism 2).
There is no curative treatment. The goal of treatment is to maximize visual function and manage complications.
Visual Management
Refractive correction: The basis is eyeglass correction from infancy. Early intervention to prevent amblyopia is important.
Tinted glasses: Use lenses that block about 20% of light indoors and about 80% outdoors 5).
Iris contact lenses: Can be used for cosmetic improvement and reduction of photophobia.
Low vision care: Utilize assistive devices such as magnifiers, video magnifiers, and tablet devices.
Nystagmus and Strabismus
Strabismus surgery: Performed for cosmetic purposes. In some cases, Dawson-Trick-Litzkow (DTL) surgery may be performed to reduce nystagmus.
Nystagmus treatment: There is no curative treatment. Strabismus surgery may be considered to address abnormal head posture (null zone).
Skin and Systemic Management
Sun protection: Skin protection with UVB-blocking sunscreen, clothing, and hats. Even in Japanese individuals, attention to the risk of squamous cell carcinoma is needed.
HPS management: NSAIDs (including aspirin) are generally contraindicated because they worsen platelet dysfunction. Regular pulmonary function monitoring from the teenage years is recommended 4).
Genetic counseling: Essential for family planning, carrier diagnosis, and subtype determination.
Antifibrotic agents are used for pulmonary fibrosis associated with HPS.
Liu et al. (2025) reported that nintedanib stabilized pulmonary fibrosis for 18 months in an HPS case with a novel homozygous HPS4 mutation 4). Pirfenidone is also a treatment option used similarly.
QIs gene therapy for albinism clinically available?
A
Currently, gene therapy for albinism is not clinically available. Gene therapy has been approved for other inherited retinal diseases such as retinoschisis and LCA (e.g., Luxturna), and research on its application to albinism is ongoing 6). Currently, refractive correction and low vision care are the mainstays of standard treatment.
Melanin synthesis proceeds within melanosomes via the following pathway:
Tyrosine → L-DOPA → DOPA quinone → (eumelanin or pheomelanin)
The conversion of dopachrome to DHICA (5,6-dihydroxyindole-2-carboxylic acid) is catalyzed by DCT (TYRP2) 2), an essential reaction for eumelanin synthesis.
OCA2 melanosome maturation defect: Reduced function of the OCA2 protein impairs the transition of melanosomes to stage IV (mature melanosomes), increasing the number of immature stage I/II melanosomes 1). Disruption of pH regulation via chloride current control is the main mechanism 1).
When pigmentation of the retinal pigment epithelium is inadequate, the light-shielding effect during development is lost, hindering the morphological development of the fovea (formation of the avascular zone and dense packing of cone cells). This is the fundamental cause of poor vision in albinism.
In albinism, fibers from the temporal retina cross excessively to the contralateral side at the optic chiasm, so that more fibers than usual are processed as nasal fibers. This misrouting is detected by VEP as asymmetry of the chiasmal coefficient 2).
In their 2025 OCA8 study, Rateaux et al. used a mouse model in which DCT mutation reduces L-DOPA to 50% of wild-type levels and showed that L-DOPA supplementation restores ophthalmic abnormalities 2). This suggests a role for L-DOPA as a melanin synthesis intermediate in optic pathway formation.
GPR143 mutation (OA1): Loss of function of the GPR143 protein impairs melanosome vesicle transport, leading to the formation of macromelanosomes 3).
HPS (BLOC complex disorders): Loss of function of BLOC-3 impairs activation of Rab32/38, resulting in general disruption of biogenesis of melanosome-related organelles (LROs: lysosome-related organelles) 4). Dense bodies in platelets and lamellar bodies in type II pulmonary epithelial cells are affected. Accumulation of ceroid-like substances from abnormal lamellar bodies is thought to be involved in the background of pulmonary fibrosis 4).
BLOC-1 (HPS-11): Functions as an eight-subunit complex including BLOC1S5 (BLOS3) and regulates endosomal recycling 5).
7. Latest Research and Future Perspectives (Investigational Stage)
L-DOPA is an intermediate in melanin synthesis and also plays an important role in retinal development and optic pathway formation.
In the OCA8 mouse model by Rateaux et al. (2025), reduced L-DOPA due to DCT mutation was the main cause of the ocular phenotype, and L-DOPA supplementation improved the abnormalities 2). Application to humans is being studied 6).
Evaluation of visual function improvement by levodopa administration in patients with albinism is ongoing, and a retinal development-promoting effect is expected 6).
Nitisinone, a 4-hydroxyphenylpyruvate dioxygenase (4-HPPD) inhibitor, is being studied for its potential to increase ocular melanin by modulating the tyrosine metabolism pathway 6).
Basic research on gene replacement therapy targeting OCA-related genes (TYR, OCA2, etc.) is progressing. The approval of Luxturna (RPE65 gene replacement) for LCA (Leber congenital amaurosis) is accelerating gene therapy research for inherited retinal diseases in general 6).
Jiang et al. (2024) identified a novel compound heterozygous mutation (c.635A>G/c.2359+1G>T) in the OCA2 gene by whole-exome sequencing (WES) in a Chinese family 1). Functional analysis confirmed that the mutant protein has impaired translocation to melanocytes compared to the wild type.
Boeckelmann et al. (2021) detailed the clinical spectrum of HPS-11 caused by BLOC1S5 mutations in a study of 5 cases, including those reported after 2020 5). Guidelines for monitoring pulmonary, ocular, and neurological symptoms have been provided.
The efficacy of antifibrotic therapy with nintedanib and pirfenidone for HPS pulmonary fibrosis is being continuously evaluated 4).
Flynn et al. (2025) reported that in fundus examinations of female OA1 carriers with GPR143 mutations, 94% had arcuate prepapillary patchy pigment changes and 74% had iris transillumination 3). It was shown that ocular abnormalities are frequently observed even in carriers.
QAre patients with albinism at high risk for skin cancer?
A
In OCA, due to the lack of skin pigment, DNA damage from ultraviolet radiation accumulates easily, significantly increasing the risk of skin cancers such as squamous cell carcinoma. The risk is considered particularly high in African OCA patients. Regular dermatological visits and thorough sun protection (sunscreen, protective clothing) are recommended.
Jiang B, Zhang H, Kan Y, Gao X, Du Z, Liu Q. Novel compound heterozygous mutations in OCA2 gene were identified in a Chinese family with oculocutaneous albinism. Mol Genet Genomic Med. 2024;12:e2297. doi:10.1002/mgg3.2297. PMID:37882226; PMCID:PMC10767448.
Rateaux M, Hadj-Rabia S, Barrois R, Zambrowski O, Michaud V, Moreno-Artero E, et al. Chiasmal Decussation in Oculo-Cutaneous Albinism Type 8. Investigative ophthalmology & visual science. 2025;66(2):44. doi:10.1167/iovs.66.2.44. PMID:39951296; PMCID:PMC11824501.
Flynn E, Cheela I, Kaden TR. Female Carrier of Ocular Albinism Linked to Gpr143 Gene. Journal of vitreoretinal diseases. 2025. doi:10.1177/24741264251386389. PMID:41180145; PMCID:PMC12578608.
Liu Q, Qing W, Guo S, Wang Y, Chen Y, Liu J. A novel homozygous HPS4 mutation in Hermansky-Pudlak syndrome: case report and literature review. Therapeutic advances in respiratory disease. 2025;19:17534666251383677. doi:10.1177/17534666251383677. PMID:41277635; PMCID:PMC12644420.
Boeckelmann D, Wolter M, Kasmann-Kellner B, Koehler U, Schieber-Nakamura L, Zieger B. A novel likely pathogenic variant in the BLOC1S5 gene associated with Hermansky-Pudlak syndrome type 11 and an overview of human BLOC-1 deficiencies. Cells. 2021;10:2630. doi:10.3390/cells10102630. PMID:34685610; PMCID:PMC8533863.
Gurnani B, et al. Nystagmus review. Clin Ophthalmol. 2025;19:1617-1648.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.